Introduction
This series of articles is intended to provide a general overview of the issues surrounding genotoxic impurities (GTIs) and their potential resolution. Part 1 will focus on general and regulatory aspects, part 2 will review toxicological issues, whereas part 3 addresses chemistry/ pharmaceutical considerations.
The presence of drug-substance and drug-product impurities that are, or may be DNA-reactive, has posed significant problems for drug regulators and industry alike over the last decade or so. The principal concern relates to drug safety in that exposure, particularly if prolonged, to compounds that can alter (alkylate) DNA may ultimately produce a carcinogenic response. A further practical issue is that the conventional testing procedures applied to drug substances for carcinogenicity and mutagenicity/genotoxicity (the property of being able to damage cellular DNA and induce genetic mutation) generally lack sufficient sensitivity to detect potentially adverse effects associated with DNA-reactive impurities (which are often present only at ppm levels).
There is considerable confusion over terminology in relation to mutagenicity and genotoxicity. It is normally accepted that “genotoxicity” is a more general term which, according to ICH guidance on genotoxicity testing [ICH S2 (R1)], can be defined as: “a broad term that refers to any deleterious change in the genetic material regardless of the mechanism by which the change is induced [1].” More detailed definitions of “mutagenicity” and “genotoxicity” (see Definitions section) are available and it is clear that the former is a more specific phenomenon applying to the induction of permanent transmissible changes in the amount or structure of the genetic material of cells or organisms, which is considered to be much more appropriate than the more general term “genotoxicity” in relation to DNA-reactive impurities. Unfortunately, the two terms are often used interchangeably and the published regulatory guidance has referred to “genotoxic impurities” and so this apparent misnomer has become established as part of the terminology applied to DNA-reactive impurities.
Regulatory Background
In December 2002, the Safety Working Party (SWP) of the European Committee for Proprietary Medicinal Products (CPMP) – now called the CHMP (Committee for Medicinal Products for Human Use), published a “Position paper on limits of genotoxic impurities” [2]. The main stated reason was to fill a perceived gap in the ICH guidance relating to impurities in drug substances and drug products (ICH Q3A/B (R2) [3,4]) particularly in relation to “those potential impurities that are expected to be unusually potent”, especially those that might not be detected using analytical methods/detection limits aligned with conventional impurity identification thresholds (i.e. in the range 100-1000ppm). Genotoxic impurities (GTIs) are considered to fulfill this potency criterion. Prior to the release of the position paper, issues concerning genotoxic impurities had been addressed on an ad-hoc basis, one example being residues of the alkylating agent 1,4-butane sultone in the excipient, sulfobutylether betacyclodextrin sodium (SBECD). This excipient is employed as part of the formulation of Vfend (voriconazole) and concerns were raised in 2001 as part of the EU assessment process about the toxic potential of this alkylating agent. Ultimately, setting a limit of < 1 ppm 1,4-butane sultone in SBECD was considered sufficient to enable the authorization of Vfend (positive opinion 13.12.2001)[5].
Around the same time the European Directorate for Quality of Medicines and HealthCare (EDQM) hypothesized that alkyl mesylate (mesilate or methanesulfonate) impurities could be present in mesylatesalt drug substances, and requested additional information on the requirements for limit tests to be included in the European Pharmacopoeia (Ph.Eur.) [6]. In 2005, the Ph.Eur. decided against setting specific limits and instead proceeded on the basis of a “production statement” for inclusion in all active pharmaceutical ingredient (API) monographs that contained mesylate salts, dihydroergocristine mesilate for example [7]. The production statement specified that: “The production method must be evaluated to determine the potential for formation of alkyl mesilates, which is particularly likely to occur if the reaction medium contains lower alcohols. Where necessary, the production method is validated todemonstrate that alkyl mesilates are not detected in the final product.” In 2011, updating proposals were released supporting the continued use of the production statement for controlling alkyl mesilates (specifically methyl, ethyl and isopropyl mesilates) in mesilate salts [8]. However, the notion that alkyl mesilates are actually formed as by-products during mesilate salt synthesis by ester formation between an alcohol solvent and methanesulfonic acid (MSA) has been challenged, since the available published data indicate that protonation of the base-form of the drug substance is effectively instantaneous [9]. If this evidence-based assessment is accepted by the European Pharmacopoeia Commission, the production statement would be redundant, although GMP requirements would remain in order to ensure that impurities in MSA (such as methanesulfonyl chloride), that can react with alcohols to produce alkyl mesylates, are maintained at suitably low levels [9].
Following the release of the CPMP Position Paper mentioned above, draft guidance was prepared, and after an extended period of consultation, an EU guideline on limits of genotoxic impurities was finally issued in 2006, with an effective date of 01st January 2007 [10].
The guideline has been subsequently updated by several Q&A supplements [11] in order to provide clarification and address some emerging issues. Consideration of issues addressed in the Q&A supplements is included in the following summary since it illustrates how regulatory thinking has evolved and how the various themes have impacted on US FDA (Food and Drug Administration) and ICH M7 deliberations.
EU Guidance
Decision Tree: ALARP/TTC (As Low as Reasonably Practicable/Threshold of Toxicological Concern)
The EU guideline on limits of genotoxic impurities includes a decision tree for the evaluation of genotoxic impurities (see scheme 1). This decision tree indicates that if there is sufficient evidence of a thresholded mechanism for genotoxicity, then a permitted daily exposure (PDE) value can be determined to ascertain safe exposure levels; this process is aligned with the approach used in ICH Q3C (R5) [12] for controlling levels of residual solvents. By contrast, for GTIs which are presumed to be non-thresholded then the applicant needs to confirm that the presence of the impurity is unavoidable (or eliminate from synthesis), show (experimentally) that the levels are as low as reasonably practicable i.e., ALARP (or reduce to ALARP levels) and finally to apply the TTC (threshold of toxicological concern) approach. If the GTI concentration is equivalent to a patient exposure (applying the MDD (maximum daily dose) of the drug substance) less than the current TTC value (1.5μg/day) then the risk of a carcinogenic response is considered negligible, and the ALARP principle does not apply. On the other hand, if the TTC is exceeded, the applicant is required to either restrict the use of the batch or reject the affected batch of API. Thus the approach could be summarized as: avoid > reduce > control (TTC).
Many Industry observers have commented that since reactive intermediates are required to achieve the efficient synthesis of APIs, particularly those that are complex and contain multiple functional groups, complete elimination of GTIs is not a practical proposition. Although, it may be possible to re-design the synthesis to avoid a specified GTI the alternative synthesis will in all probability have other GTI(s) that would also need to be controlled. The use of less reactive reagents might be considered as a partial solution since any residues in the API would most likely have reduced DNA-reactivity. However, this strategy is considered inappropriate since synthetic yields would probably be reduced and any reagent residues would be less likely to be deactivated by downstream processing, and be more environmentally persistent (‘green chemistry’ considerations).
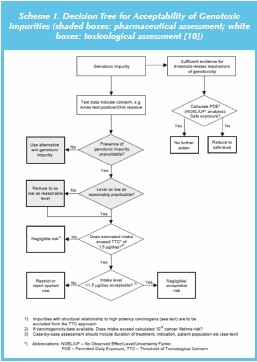
The consolidated Question and Answer (Q&A) supplements [11] indicate that there is no cause for concern (in terms of GTIs) for synthetic-processes that remain unchanged (from before implementation of the guideline) and in the absence of new knowledge no re-evaluation of the synthetic route is normally required. One cited example of new knowledge is a switch to a sulfonic acid salt API, accompanied by the potential for formation of genotoxic alkyl sulfonates, although such concerns now seem overstated in light of the recent evidence-based evaluation of alkyl mesylates discussed above [9].
Pharmaceutical Issues
No action is required for an unidentified impurity if the impurity is below the ICH identification threshold. Identified impurities present at a concentration above the ICH identification threshold, but below the qualification threshold, need to be evaluated in silico and/or in an Ames assay if a structural alert is present. Alternatively, Ames-test data may be available in the public domain on the impurity as such or on one or more close structural analogues. A negative outcome to these evaluations will remove any concerns. If the structure cannot be elucidated, no further action would be required. Ames-negative structurally alerting impurities at concentrations exceeding the ICH Q3A/B qualification thresholds [3,4] would still need to be evaluated using the Q3A/B guidance, possibly involving the normal suite of genotoxicity/subchronic toxicity studies.
For established drug substances (marketed prior to the issuance of the guideline), either with or without a pharmacopoeial monograph, the presence of an impurity showing structural alerts alone is insufficient to justify additional measures (such as Ames testing), unless there are additional data demonstrating a cause for concern. If changes are madeto the synthetic route that could generate different GTIs or higher levels of established GTIs, then these issues should be discussed with the competent authority.
PGIs (potential genotoxic impurities) that might be formed based on theoretical considerations, but which are not found in practice, do not need to be included in the API specification. If a PGI were to be present at the penultimate stage of an API synthesis, and if it could be shown that this impurity can be controlled by in-process limits and if the impurity does not exceed 30% of the proposed limit (TTC or other acceptable limit) in the API (based on data from spiking experiments), exclusion of this PGI from the drug substance specification should be possible. For those cases where a PGI (reagent, degradation product or intermediate) occurs in the final synthetic stage, the impurity should be included in the API specification (unless shown to be Ames-negative or otherwise qualified). Skip-lot testing can be applied to the PGI if it does not exceed 30% of the proposed limit in the API. Impurity profile data from 6 consecutive pilotscale batches or 3 consecutive production-scale batches are required to support skip-testing; otherwise routine testing needs to be applied [12].
Structural Alerts
A negative Ames test over-rules a structural alert provided that the GTI remains below ICH Q3A/Q3B levels [2,3]. The absence of a structural alert emerging from a well performed assessment using QSAR (quantitative structure activity relationship) software such as Derek Nexus [13] or MULTICASE [14] is considered sufficient evidence to justify performing no additional qualification studies. For compounds with a structural alert, it is permissible to control such an impurity to levels less than the TTC, without resorting to Ames-testing provided that the structural alert is not of high concern (N-nitroso, aflatoxin-like, etc) [11].
Staged TTC
The staged TTC concept, proposed by Mueller et al. [15] and based on an earlier publication by Bos et al. [16] on the virtually safe dose (VSD), can be used during clinical development and marketing. This concept takes into account the fact that duration of exposure is a key factor impacting on the probability of a carcinogenic response. The recommended limits for daily intake of GTIs are: 1.5, 5, 10, 20 and 60μg/day for greater than 12-months, 6-12-months, 3-6-months, 1-3-months and less than 1-month, respectively. For single doses, an intake of 120μg/day is acceptable [11].
Multiple GTIs
The TTC level can be applied singly to individual GTIs only if the impurities are not structurally related. For GTIs that are structurally similar, it is assumed that they could have the same mode of action and attack the same molecular target, and thus their effects might be additive in nature. EMA therefore recommended that the appropriate individual limit (default TTC or staged-TTC) should be applied to the sum of structurally similar GTIs [11].
However, Elder and Harvey [17] argue that the concept of additive risks proposed by regulatory agencies is supported mainly by assertion rather than evidence. According to Elder and Harvey, recent modeling studies indicate that there is a slightly increased cancer risk arising from multiple GTIs (>3), but that it is insignificant compared to the extremely conservative assumptions incorporated into the TTC. These authors suggested that there are serious difficulties associated with defining both ‘common mechanisms of toxicity’ and ‘concurrent exposure’. When taken together with the limited data and associated methodologies that are currently available to conduct cumulative risk assessments, it is believed that the concept of applying a group TTC to structurally similar GTIs is not well established.
In addition, there are significant analytical challenges inherent in assaying and controlling multiple GTIs to levels below the TTC (potentially ppb limits).
Herbal Products
The other area where European GTI guidance has been issued is with respect to herbal products [18]. The EMA Committee on Herbal Medicines Product (HMPC) released a concept paper proposing the development of guidelines on genotoxic impurities in herbal substances and preparations. HPMC indicated that the contribution of the substance to the overall exposure needs to be considered, as herbal products are made of natural substances that may form part of a regular, dietary and/ or environmental exposure.
HMPC emphasized the requirement for rugged risk-benefit assessments on herbal products, conceding that the complex, as well as variable nature of herbal products (season-to-season, geographical origins, or preparation process), present different challenges compared to those from conventional medicinal products. HMPC further indicated that “the complete composition is very difficult to unravel, so one can argue that there are always many unknown constituents and thus there may be ‘hidden’ dangers.”
In its final guidance [18], HPMC has included a decision tree to evaluate the potential risks inherent in a herbal remedy. HPMC stressed that since herbal remedies are complex mixtures, closely following these requirements may pose technical difficulties for reliable genotoxicity assessments.
Draft FDA Guidance
In 2008 the US FDA (Food and Drug Administration) draft guidance on genotoxic and carcinogenic impurities, was made available for public consultation [19].
There are marked similarities [20] with the EU guideline, in terms of TTC, the acceptance of a staged approach where the duration of treatment is limited (during drug development) and the use of SAR (structure activity relationships) as part of the evaluation. However, there are some significant differences, notably the requirement to introduce lower limits for vulnerable patient populations, especially pediatrics. Also there appears to be a greater level of flexibility regarding the avoidance>ALARP>TTC hierarchy. There are minor differences in the staged TTC as applied to first into man studies of less than 14 days, where FDA favored the extension of the 120μg/day limit for the total duration of this period. In addition, the FDA draft guidance does not allow the application of the staged TTC to marketed products.
In 2011 an expert group began working on a new ICH guideline, M7 [21], on genotoxic impurities. Based on the Step 1 version of M7 that has been distributed on a restricted basis it is apparent that the key principles of the guidance are likely to reproduce those enunciated in the EU guideline (and in the FDA draft guidance, excluding carcinogenic impurities). When finalized, ICH M7 will supersede both the EU guideline [10, 11] and the draft FDA guidance [19].
ICH M7 (Step 1)
The purpose of ICH M7 (step 1) [21] is to provide a practical framework that can be applied for the identification, categorization, qualification, and control of mutagenic impurities to limit potential carcinogenic risk. In terms of scope, the API categories mentioned in ICH Q3A/B [3,4] are also excluded from guidance on genotoxic impurities: biological/ biotechnological, peptide, oligonucleotide, radiopharmaceutical, fermentation and herbal products, and crude products of animal or plant origin. Impurities in products used in advanced cancer indications (see ICH S9 [22]) and impurities in a drug substance that is it itself genotoxic are also excluded. The limits described are applicable to all routes of administration and all patient populations.
As with earlier guidance, the ICH M7 (step 1) guideline [21] is intended to provide guidance for marketed registration applications, as well as clinical trial products. It will also be applicable to new amendments of existing authorizations, where changes to the API synthetic route may result in higher specified levels of existing impurities or new impurities (only the impacted impurities requiring evaluation). Formulation changes will be in-scope if the changes result in either higher specified levels of existing impurities/degradants or new impurities/degradants (only the impacted impurities/degradants needing evaluation). In addition, new applications for previously approved products producing changes in patient population, therapeutic indication(s), or dosing regimen, will be covered bythe guidance if the change significantly impacts on the acceptable cancer risk.
The general principles enunciated in the Step 1 ICH M7 [21] guideline parallel those in the existing EU guidance [10, 11], particularly in relation to structural alerts, Ames testing, thresholded and non-thresholded compounds and application of the TTC concept. The guideline also advocates the use of risk assessments. Thus, in addition to the known impurities defined in drug substance specifications, the applicant should assess PGIs, which may be present at levels below the identification thresholds defined in ICH Q3A/3B [3,4]. In addition to API impurities, the guideline recommends assessment of potential degradants in the drug product. This can be based on knowledge of known relevant degradation pathways using standard ICH stress conditions, i.e. 40°C/75%RH for 6-months or photostability studies described in ICH Q1B [23].
The initial assessment is to determine if a particular impurity contains one or more electrophilic centers that might constitute structural alerts for DNA-reactivity (mutagenicity). If an alert is identified, an Ames test can be undertaken to follow up a structure-based concern to identify impurities that are DNA-reactive mutagens with carcinogenic potential. Thus, a structural alert would classify an impurity as a PGI, whereas the classification of an impurity as “genotoxic” would mean that there are “positive findings in established in vitro and/or in vivo genotoxicity tests”. ICH M7 [21] also indicates that the focus of the guidance is on DNA-reactive substances that have a potential to directly cause DNA damage when present at low levels leading to mutations and therefore can potentially cause cancer. This type of mutagenic carcinogen is usually detected in a bacterial reverse mutation test (Ames test). In other words, somewhat confusingly, the guidance suggests that there may be a cause for concern in relation to impurities that are “genotoxic” in general, whereas the focus is on impurities that test positive in bacterial reverse mutation assays.
In terms of identifying structural alerts and predicting the outcome of appropriate genotoxicity assays, ICH M7 states: “Structure-based assessments are useful for predicting Ames outcomes based upon the established knowledge base. There are a variety of approaches to conduct this evaluation including a review of the available literature, expert knowledge, and/or computational toxicology assessment.”
As previously mentioned in the earlier EU guideline [10], genotoxic impurities are divided into two categories:
- Genotoxic compounds with sufficient (experimental) evidence for a threshold-related mechanism;
- Genotoxic compounds without sufficient (experimental) evidence for a threshold-related mechanism.
For the former (for example, topoisomerase inhibitors and most intercalating agents), acceptable limits can be obtained by determination of a PDE (permitted daily exposure) value based on an experimental NOEL (no observed effect level) using ICH Q3C (R5) [12] methodology (as applied to residual solvents).
For the latter group of non-thresholded compounds, which are likely to be in the majority, experimental demonstration of an in-vivo threshold is unlikely to be straightforward and no agreed and validated procedures appear to be available for such purposes. Consequently, as a pragmatic solution, the concept of the Threshold of Toxicological Concern - TTC (in relation to carcinogenic activity) has been adopted. The current default value for the TTC is 1.5 μg/day. This corresponds to a lifetime risk of cancer of 1 in 100,000, based on an evaluation by Kroes et al. [24] of carcinogenic potency data on 730 carcinogens selected from the CPDB (Carcinogenic Potency Database) [25]. Kroes et al. [24] also identified a “cohort of concern” comprising high-potency genotoxic carcinogens such as aflatoxin-like compounds, N-nitroso and azoxy compounds, that should be excluded from the default TTC limit. No general limit for these high-potency compounds was recommended, although 0.15 μg/day might be considered acceptable by some toxicologists. There is a high level of conservatism built into the derivation of the standard TTC limit and this is acknowledged to some extent in the regulatory guidance.
The TTC limit is applied to impurities that are DNA-reactive (based on positive results in bacterial reverse mutation assays) and to structurally alerting impurities for which no mutagenicity data are available. There are a number of exceptions however, which include:
• Impurities for which carcinogenicity bioassay data are available; the TTC should not be applied in these cases; rather, a compound-specific risk assessment can be made by linear extrapolation of carcinogenicity potency data using a risk value of 1 in 105.
• Less than lifetime (LTL) exposure for which a “staged-TTC limit”, ranging from 120-10 μg/day (based on the sum of all genotoxic impurities if > 3 GTIs are present), can be applied to marketed products or those in clinical development.
• Compound class-specific acceptable limits higher than the default TTC can be applied based upon the recognized differences in carcinogenic potencies associated with different structural alert categories, e.g. mono-functional alkyl halides
• Higher limits may be justified when human exposure to the impurity will be much greater from other sources e.g., food, one example being crotonaldehyde (2-butenal) [26].
• Case-by-case exceptions to the use of the appropriate TTC can be justified in cases of severe disease, reduced life expectancy, or with limited therapeutic alternatives.
When up to three genotoxic impurities are present the individual TTC limit can be applied to each impurity.
The guideline is not intended to be applied to precedented excipients, where there are decades of safe usage in marketed products [27]. Similarly, the guidance does not apply to leachables arising from drug product container/closures; however, the principles included in the guidance can be applied, if warranted, in the management of carcinogenic risk.
Critical Assessment of ICH M7 Regulatory Principles
Many evaluations have been made of the ability of in-vitro genotoxicity tests to predict carcinogenic activity. In this regard, the Ames assay is reported to have a positive/negative predictivity of approximately 80% and 50% respectively [28]. The negative predictivity reflects the quite high probability of Ames-negative compounds exhibiting some kind of carcinogenic response (i.e. non-genotoxic carcinogens) which is explained mainly by the use of extremely high doses in carcinogenicity bioassays (maximum tolerated dose) leading to cytotoxicity and metabolic overload. Consequently, the presumption that an Ames-positive impurity might well have carcinogenic activity is not unreasonable, although a sizeable number of mutagenic compounds have tested negative in carcinogenicity bioassays [29].
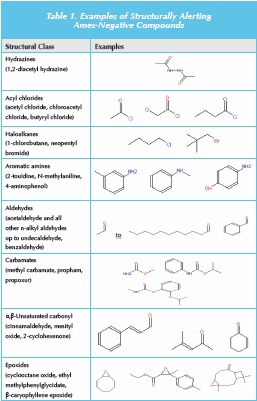
What about the implied correlation between a structural alert and carcinogenic activity (if no genotoxicity data are available)? Firstly, it is worth defining what is meant by a structural alert. This term was originally proposed by Ashby and Tennant [30] based on the good structure mutagenicity (in Salmonella)/carcinogenicity correlation for compounds containing an electrophilic functional group (or a compound that could be metabolised to one containing such a reactive entity, e.g. some aromatic amines). Figure 1 shows a listing of functional groups identified as structural alerts using Ashby-Tennant criteria [31]. Other listings may widen the scope of some alerts [32], for example including all aldehydes rather than just alkyl aldehydes (and possibly including other genotoxicity endpoints such as clastogenicity). The link between a structural alert and carcinogenic potential is much more tenuous than that for Ames-positive compounds, and highly dependent on the particular alert. For example, all simple n-alkyl aldehydes, except formaldehyde, are Ames-negative [33], and for carbamates, only vinyl carbamates or compounds that can be metabolised to vinyl carbamates, are genotoxic [34]. Other examples are shown in Table 1. Thus, there is considered to be a lack of clarity on structural alerts, and a more detailed description of the various alerts, and likely sequelae in terms of carcinogenicity, need to be included in the guidance.

Although ICH M7 [21] stresses that the guidance is focused on DNA reactive impurities, and that other types of genotoxic compounds that are Ames-negative have thresholded mechanisms, there remains a degree of ambiguity regarding Ames-negative impurities that show positive results in mammalian-cell assays (such as the mouse lymphoma assay or in-vitro cytogenetic assays). This issue has led to markedly differing interpretations of the current EU guidance and it is a disappointment that the (current) wording of ICH M7 still leaves open the option of classifying Ames-negative, mammalian-cell positive compounds as “genotoxic impurities”.
The treatment of multiple genotoxic impurities (GTIs) is rather unclear especially in terms of the recommended limits if > 3 GTIs are present. Will the limit be 3/n x 4.5 μg/day (where n = number of genotoxic impurities), or something different? Concerning the TTC applied to GTIs and some PGIs, many may be unaware that the TTC is a general concept in that there is not just one unique TTC, but many. In fact the structure-based TTCs for non-cancer endpoints are used extensively in the evaluation of food flavoring substances by the Joint Expert Committee on Food Additives (JECFA) and the European Food Safety Authority (EFSA) [35].
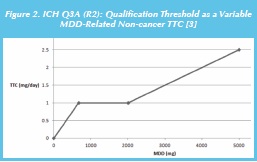
Several more recent independent assessments, such as that by Tluczkiewicz et al. [36] using different data sets have produced noncancer TTCs that are remarkably similar to those originally proposed by Munro et al. in 1996 [37]. A further consideration is that the qualification thresholds specified in ICH Q3A/B (R2) are, in effect, equivalent to MDD (maximum daily dose)-dependent non-cancer TTCs as illustrated in Figure 2 for ICH Q3A (R2) [3]. At a drug substance MDD of 667-2000 mg, the non-cancer TTC is 1 mg, although the TTC is lower with MDD < 667 mg and higher with MDD > 2000 mg.
A recent detailed assessment [29] has highlighted the extremely over-conservative and potentially biased nature of the derivation of the TTC as applied to genotoxic impurities, and a re-evaluation is advocated in order to provide a sound scientific basis for ICH M7.
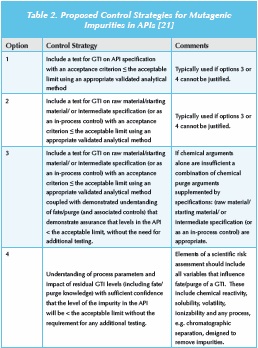
Finally, the guideline advocates the development of a control strategy for the drug substance. Four control strategies are defined for API impurities, which are summarized in Table 2. The guidance is less prescriptive for drug product degradants and highlights the need for good formulation/packaging development to minimize exposure to moisture, light or oxygen. However, if these approaches fail to control the genotoxic degradant to below an appropriate acceptance criterion, then a risk/benefit evaluation will be required to assess the appropriate control strategy. The guidance indicates that both avoidance and ALARP are unnecessary as long as the GTI levels are below an acceptable limit.
Pharmaceial Considerations
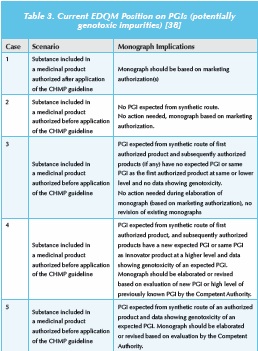
The European Directorate for Quality in Medicines and Healthcare (EDQM) [38] have also identified the requirement to develop a process for dealing with potentially genotoxic impurities (PGIs), that can be applied during the review and revision of pharmacopeial monographs. They propose the adoption of a pragmatic approach for existing monographs, and argue that in the absence of new data demonstrating the genotoxicity of a PGI, then structural alerts alone are insufficient to trigger any additional measures. Thus, retrospective application to marketed products (containing an API from a marketed source) is considered unnecessary, which is aligned to the regulatory expectations within Europe. A recent presentation from EDQM [39], provided further guidance and indicated that the ’transparency lists’ of known impurities in pharmacopoeial monographs may contain alerting structures. However, these structural alerts do not automatically imply genotoxicity. A production statement may be included to flag PGIs, although this provision is currently limited to monographs for mesilate-salt APIs. These transparency lists of “other detectable impurities” are for analytical information only and do not necessarily confi rm their occurrence at relevant levels since Ph. Eur. monographs may relate to several routes of synthesis and the designated impurity/PGI may not be relevant for a particular routes of synthesis.
EDQM have summarized their position (see Table 3) and this provides useful insight into the current pharmacopeial position on PGIs in Europe. Monographs (limit and tests) need to be updated if relevant information is submitted from stakeholders (in particular National Competent Authorities). Importantly, the existence/use of a monograph does not release the user from his responsibility to review the synthetic route, the process control and the impurity profile as regards PGIs. In addition, revision of the general monograph 2034 on substances for pharmaceutical use has been proposed in terms of applying the EMA guideline on limits of genotoxic impurities: “When active substances are included in a new application for a medicinal product for human use, the requirements of the guideline on the limits of genotoxic impurities and the corresponding questions and answers documents published on the website of the European Medicines Agency (or similar evaluation principles for non-European Union member states) must be followed” [40]. The USP has held similar discussions on issues relating to PGIs [41].
Conclusion
Overall, the current regulatory and pharmacopeial approaches are considered to exhibit a reasonable level of pragmatism, but some of the underlying principles often appear to lack clarity and precision, for example, regarding ‘common mechanisms of toxicity’, analysis of structurally similar GTIs and Ames-negative compounds with one or more positive results in mammalian-cell genotoxicity assays. It is to be hoped that a dialogue will be established around ICH M7 leading ultimately to a guideline that is acceptable to Industry and regulatory authorities alike.
Definitions
- Potentially genotoxic impurity (PgI): compound with structural alert.
- Mutagenicity refers to the induction of permanent transmissible changes in the amount or structure of the genetic material of cells or organisms. These changes may involve a single gene or gene segment, a block of genes or chromosomes. The term clastogenicity is used for agents giving rise to structural chromosome aberrations. A clastogen can cause breaks in chromosomes that result in the loss or rearrangements of chromosome segments. Aneugenicity (aneuploidy induction) refers to the effects of agents that give rise to a change (gain or loss) in chromosome number in cells. An aneugen can cause loss or gain of chromosomes resulting in cells that have not an exact multiple of the haploid number. For example, three number 21 chromosomes or trisomy 21 (characteristic of Down syndrome) is a form of aneuploidy [42].
- Genotoxicity is a broader term and refers to processes which alter the structure, information content or segregation of DNA and are not necessarily associated with mutagenicity. Thus, tests for genotoxicity include tests which provide an indication of induced damage to DNA (but not direct evidence of mutation) via effects such as unscheduled DNA synthesis (UDS), sister chromatid exchange (SCE), DNA strandbreaks, DNA adduct formation or mitotic recombination, as well as tests for mutagenicity [42].
- Clastogenic: Interaction with chromosomes.
- Mutation: induced, transferable change of the cell‘s genetic material.
- Carcinogenicity: the ability or tendency to produce cancer (longterm studies).
References
1. ICH S2(R1), Guidance on Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended for Clinical Use, Current step 4 version 1, 09th November 2011: http://www.ema.europa. eu/docs/en_GB/document_library/Scientific_guideline/2011/12/ WC500119604.pdf. Accessed on 1st May 2012.
2. D. Kirkland, D. Snodin. Setting limits for genotoxic impurities in drug substances: threshold-based and pragmatic approaches. Int. J. Pharm Med., 18, 2004, 197-207.
3. ICH Q3A, Impurities in New Drug Substances, in International Conference on Harmonization Tripartite Guideline. Current Step 4 version 2. June 2008.
4. ICH Q3B, Impurities in New Drug Products, in International Conference on Harmonization Tripartite Guideline. Current Step 5 version 1. June 2006.
5. Vfend (voriconazole) EPAR: http://www.ema.europa.eu/ docs/en_GB/document_library/EPAR_-_Scientific_Discussion/ human/000387/WC500049753.pdf. Accessed on 23 April 2012.
6. European Directorate for Quality of Medicines and Healthcare. Enquiry: alky mesilate (methanesulfonate) impurities in mesilate salts, Pharmeuropa, 12, 2000, 27.
7. European Pharmacopoeia. Dihydroergocristine mesilate monograph, Ph. Eur., 7.0, 2012.
8. Review of the Production statement in monographs for mesilate salts. Pharmeuropa 23.4, 2011, 691-692.
9. D.J. Snodin, A.C. Cartwright, B.R. Matthews. Production statement for mesilate-salt drug substances is no longer justifi ed. Pharmeuropa Readers’ Tribune, April 2012, 1-10.
10. Committee for Medicinal Products for Human Use (CHMP), Guideline on the Limits of Genotoxic Impurities, 01 January 2007.
11. ICH Q3C, Guideline for Residual Solvents, in International Conference on Harmonization Tripartite Guideline. Current Step 4 version 2. 04th February 2011.
12. European Medicines Agency, Questions and Answers on the ‘Guideline on the Limits of Genotoxic Impurities’, EMA/CHMP/ SWP/431994/2007 revision 3, 23rd September 2010. Accessed on 23rd April 2012.
13. Derek Nexus. LHASA Ltd: https://www.lhasalimited.org/derek_ nexus/. Accessed 1st May 2012.
14. MULTICASE Inc Bioactivity Software: http://www.multicase. com/. Accessed 1st May 2012.
15. L. Mueller, R.J. Mauthe, C.M. Riley, M.M. Andino, D De Antonis, C. Beels, J. DeGeorge, A.K.M. De Knaep, D Ellison, J.A. Fagerland, R. Frank, B. Fritschel, S. Galloway, E. Harpur, C.D.N. Humfrey, A.S. Jacks, N. Jagota, J. MacKinnon, G. Mohan, D.K. Ness et al., A Rationale for Testing, and Controlling Specified Impurities in Pharmaceuticals that Posses the Potential for Genotoxicity, Reg. Tox. Pharm., 44, 2006, 198-211.
16. P.M. Bos, B.J. Baars, M.T. van Raaij. Risk assessment of peak exposure to genotoxic carcinogens: a pragmatic approach. Toxicol Lett. 151(1), 2004, 43-50.
17. D.P. Elder and J.S. Harvey. Is there a Real Case for Cumulati ve Control of Structurally Related Genotoxic Impurities? Org. Proc. R.D., 14, 2010, 1037-1045.
18. European Medicines Agency, Committee on Herbal Medicines Product, Guideline on the Assessment of Genotoxic Constituents in Herbal Substance /Preparations, EMEA/HMPC/107079/2007, London, 21st May 2008.
19. Guidance for Industry, Genotoxic and Carcinogenic Impurities in Drug Substances and Products: Recommended Approaches, U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), December 2008.
20. R.Ogilivie and A. Teasdale. Historic Overview of the Development of Genotoxic Impurities Guidelines and Their Impact, In: Genotoxic Impurities (strategies for Identification and Control), A. Teasdale (Editor), Wiley, New Jersey, 2010, pp. 3-26.
21. ICH M7, Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk, in International Conference on Harmonization Tripartite Guideline, Step 1 working draft, 11th November 2010.
22. ICH S9. Non Clinical Evaluation for Anticancer Pharmaceuticals. Step 4. 29th October 2009.
23. ICH Q1B (R5). Photostability Testing of New Active Substances and Medicinal Products. Step 5, J anuary 2008.
24. R. Kroes, A.G. Renwick, M.Cheeseman, J. Kleiner, I.Mangelsdorf, A.Piersma, B.Schilter, J.Schlatter, F. van Scothorst, J.G. Vos, G.Würtzen. European branch of the International Life Sciences Institute. Structure-based thresholds of toxicological concern (TTC): guidance for application to substances present at low levels in the diet. Food Technol. Toxicol. Lett., 42, 2004, 65-83.
25. CPDB Carcinogenic Potency Database: http://potency.berkeley. edu/chemnameindex.html. Accessed 2nd May 2012.
26. 2-Butenal. Concise International Chemical Assessment Document 74 (2008): http://www.inchem.org/documents/cicads/cicads/ cicad74.pdf. Accessed on 25th April 2012.
27. D.J. Brusick, A Perspective on Testing of Existing Pharmaceutical Excipients for Genotoxic Impurities, Reg. Tox. Pharm., 55, 2009, 200-204.
28. EFSA Scientific Committee. Scientific opinion on genotoxicity testing strategies applicable to food and feed safety assessment. EFSA Journal, 9(9), 2011,2379: http://www.efsa.europa.eu/en/ efsajournal/doc/2379.pdf . Accessed 1st May 2012.
29. D.J. Snodin, S.D. McCrossen. Guidelines and Pharmacopoeial Standards for Pharmaceutical Impurities: Overview and critical assessment, [in press] (http://dx.doi.org/10.1016/j. yrtph.2012.03.016). Regul. Pharmacol. Toxicol., 2012.
30. J. Ashby, R.W. Tennant. Definitive relationships among chemical structure, carcinogenicity and mutagenicity for 301 chemicals tested by the U.S. NTP. Mutat. Res. 257(3), 1991, 229-306.
31. International Programme on Chemical Safety. WHO Food Additive Series 40: http://www.inchem.org/documents/jecfa/ jecmono/v040je17.htm. Accessed on 25th April 2012.
32. K. Sawatari, Y. Nakanishi, T. Matsushima. Relationships between chemical structures and mutagenicity: a preliminary survey for a database of mutagenicity test results of new work place chemicals. Ind Health. 39(4), 2001, 341-5.
33. D. J. Snodin. Genotoxic Impurities: A regulatory toxicology commentary on recent articles in Organic Process Research Development, Org. Process Res. Dev., 2011, 15 (6), 1243–1246.
34. G.A.Dahl, J.A.Miller, E.C.Miller. Vinyl carbamate as a promutagen and a more carcinogenic analog of ethyl carbamate. Cancer Res., 38(11), 1978, 3793-3804.
35. EU Scientific Committees, Draft opinion on the threshold of toxicological concern (TTC) approach for the safety assessment of chemical substances: http://www.efsa.europa.eu/en/ consultationsclosed/call/110712a.pdf. Accessed on 25th April 2012.
36. I. Tluczkiewicz I, H.E. Buist, M.T. Martin, I. Mangelsdorf, S.E. Escher. Improvement of the Cramer classification for oral exposure using the database TTC RepDose-A strategy description. Regul. Toxicol. Pharmacol. 61(3), 2011, 340-50.
37. I.C.Munro, R.A.Ford, E.Kennepohl, J.G.Sprenger. Correlation of structural class with no-observed-effect levels: A proposal for establishing a threshold of concern. Food Chem Toxicol. 34(9), 1996, 829-67.
38. European Directorate for Quality of Medicines and Healthcare.Potential Genotoxic Impurities and European Pharmacopoeia Monographs on Substances for Human Use, Pharmeuropa, 20, 2008,426.
39. M. Wierer. Control of Impurities in the European Pharmacopoeia, Expertentag Swissmedic, Bern, 24th October 2011.
40. Substances for pharmaceutical use. Pharmeuropa. 23 No 4. October 2011. 691-693: http://pharmeuropa. edqm.eu/PharmeuropaArchives/ . Accessed 1st May 2012.
41. United States Pharmacopeia, Impact on the Future of Pharmacopoeial Standards, Session IV: Impurities in Drug Substances and Products. Genotoxic Impurities, USP Annual Scientific Meeting, Denver, Colorado, 29th September 2006.
42. EFSA Scientific Committee. Scientific opinion on genotoxicity testing strategies applicable to food and feed safety assessment. EFSA Journal 2011;9(9):2379: http://www.efsa.europa.eu/en/efsajournal/doc/2379. pdf (accessed 1st May 2012).
Dr. David Elder studied chemistry at Newcastle upon Tyne, before moving to Edinburgh University to undertake research for a Ph.D. in Crystallography. Dr. Elder has 34-years experience in the pharmaceutical industry at a variety of different companies (Sterling, Syntex and GSK). For the last 19-years he has been employed by GSK. He is currently a director in the externalization group (Scinovo) in GSK R&D. Dr. Elder is a member of the British Pharmacapoeia (Expert Advisory Group PCY: Pharmacy), a member of the Analytical Division Council (Royal Society of Chemistry, UK) and a council member of the Joint Pharmaceutical Analysis Group, UK. He is a member of the PhRMA and EfPIA sub-groups on genotoxic impurities and was part of the PQRI group that assessed the control strategies for alkyl mesilates. His recent publications are focused the chemistry/analytical evaluation of genotoxic impurities. He has authored three chapters in the recently published Genotoxic Impurities: Strategies for Identification and Control (Ed. Andrew Teasdale, Wiley, 2010.
David Snodin has been involved in regulatory toxicology for over 30 years, in both industry, and at the UK Regulatory Agency. In February 2010 he formed his own consulting business - Xiphora Biopharma Consulting. His recent publications are focused on the safety evaluation of genotoxic and non-genotoxic pharmacopoeial and other impurities.