Introduction
Analytical method transfers are an integral part of the product lifecycle of medicinal products, particularly as they are moved between different sites (often not within the same company, i.e., outsourcing). There is a requirement under current good manufacturing practice (cGMP) that methods are suitably validated and verified under actual conditions of use, which may necessitate suitable validation and method transfer to the intended site(s) of use [1].
ICH Q10 (Pharmaceutical Quality System) covers technology transfer, both of new products transferring from development into production (and supporting methodology) and transfers ‘within or between manufacturing and testing sites for marketed products’ [2]. ICH Q10 indicates that, ‘knowledge obtained during transfer and scale-up activities can be useful in further developing the control strategy’ and that CAPAs (corrective actions and preventative action) can ‘be used as an effective system for feedback, feed-forward and continual improvement.’
However, the European Medicines Agency (EMA) recently highlighted concerns that they have with respect to lack of adequate method validation specifically covering technology transfer [3]. The agency noted that this resulted in methods being cited as the primary cause during out-of-specification (OOS) investigations and that these situations frequently occur at contract Quality Control organizations (CQCO). The agency indicated that, ‘In the context of globalization of manufacturing activities and the developments of new concepts like Quality by Design (QbD), where technology transfer is expected to happen worldwide within and between companies, within the same country, as well as between countries, the need for guidance for transfer of analytical methods has been identified’ [4]. Current approaches to analytical technology transfer – such as analyzing three batches each in duplicate at the transferring and receiving sites – represent a one-off exercise aimed at proving that the receiving laboratory can operate the analytical method [5-8]. These exercises only confirm that the analyst, equipment, and other components involved in the exercise can operate the method at the time of the transfer exercise rather than providing the assurance that the method will be reliable throughout the lifecycle of its use.
Therefore, despite these methods being fully validated prior to transfer, in line with the accepted global guidance on method validation (ICH Q2 (R1)) some methods are still being found to be non-robust during routine QC testing; i.e. not fit for purpose [9]. This raises an intriguing prospect that you can successfully validate an intrinsically ‘bad’ method!
This article will explore some of the underlying reasons for this and why the application of QbD concepts can be useful in improving overall robustness of analytical methods.
ICH Q2(R1) Validation of Analytical Procedures
The ICH guidance and the related pharmacopeial guidance, i.e. United States Pharmacopeia (USP) general chapter <1225> for the development and validation of analytical methods is typically used to support a one-off validation exercise with little consideration given to verifying how well the method will perform in ‘real world’ operating conditions [9, 10]. Numerous statistics are generated (associated with method characteristics such as accuracy, precision, specificity, detection (DL / LOD) and quantitation limits (QL / LOQ), linearity and range) which satisfy the above validation guidance but are not directly linked to the method performance. Only robustness and ruggedness [11] (repeatability, intermediate precision and reproducibility) provide an insight into how the method will fare during routine use and are the only method validation characteristics which assess the impact of critical analytical factors on the method requirements (termed as the Analytical Target Profile or ATP) [12].
Method robustness is defined as, ‘a measure of its capacity to remain unaffected by small, but deliberate variations in method parameters and provides an indication of its reliability during normal usage’ [9, 10]. Robustness assesses how susceptible the methodology is to unintended variations in the defined method conditions that could be suitably anticipated to occur as a result of changing the site of analytical testing, manufacturing or both [13]. For a high performance liquid chromatography (HPLC) method, typical variations that should be evaluated are the influence of:
- Variation in temperature of method
- Variations in mobile phase composition
- Variation in pH of aqueous component of mobile phase
- Variations in flow rate of mobile phase
- Variations in detection wavelength
One outcome of robustness is (or should be) the derivation of system suitability criteria to ensure the performance of the method is satisfactory during the method lifecycle. System suitability is based on the idea that the analytical instrument (e.g. for an HPLC system this comprises of a pump, autosampler, detector and integration software) together with the analytical operations and samples comprise an integral system that can be evaluated holistically. For HPLC, these can include critical resolutions, column efficiency and peak symmetry. Typically, no guidance is provided on acceptable criteria - this is left to the authors to define. John Dolan provides some useful guidance for interested parties [14].
Method ruggedness can be defined as ’the degree of reproducibility of test results obtained by the analysis of the same sample under a variety of normal test conditions’ [10, 11]. This encompasses repeatability (same analyst/equipment over a short period of time), intermediate precision (within laboratory variation) and reproducibility (between laboratories) which are defined in the ICH guidelines under the precision validation characteristic [11]. Most companies do not identify the equipment that should be used, allowing the receiving site to select their preferred equipment; whereas, others do identify the equipment and require justification of equivalence [15] when these differ between transferring and receiving units [16]. However, the latter approach may be fundamentally flawed and it is often the first exposure to ‘different’ analytical equipment that highlights deficiencies and concerns with the methodology. Risk based approaches are preferable which can be used to proactively identify factors that can either be fixed/controlled or studied using a method ruggedness study. For an HPLC method, typical factors that should be evaluated are:
- Environmental factors associated with site of analysis (e.g. humidity)
- Analysts
- Instruments (type and configuration)
- Variations in Stationary Phases (different lots and/or suppliers)
- Samples
Non-reliable Methods
Given the extensive validation that methods undergo, why should there still be issues arising at the receiving site(s)? Interestingly, there are very few publications on the non-robust nature of analytical methods in general and yet this is a commonly reported deficiency by regulatory agencies worldwide. It may be that analytical scientists are reluctant to highlight failures and are much more likely to publish only details of methods that have been successfully validated. However, there have always been anecdotal ‘reports’ of stationary phases that were ‘less rugged’ than their peers and this has driven an evolutionary process within column manufacturers to improve the ruggedness of their stationary phases. There were particular issues with nitrile stationary phases [17] in the early 90s that resulted, in extremis, of retention time switching of critical impurities, where the relative retention times (RRTs) of these impurities could not be relied upon and the methods were nonrobust and poorly transferrable.
Inadvertently, the pharmacopeia’s approaches to the interchangeability of stationary phases have not helped this situation. The USP indicates that, ‘The specific type of packing needed to complete an analysis is indicated by the “L” designation in the individual monograph’ [13]. A complete list of packings “L”, phases “G” and supports “S” are provided in the USP. The USP also indicates that, ‘Compendial monographs do not include the name of the appropriate columns; this omission avoids the appearance of endorsement of a vendor’s product and natural changes in the marketplace’. However, even a cursory appraisal of a manufacturer’s column catalogue(s) will show the huge diversity of physical properties (e.g. particle shape/size (μm), pore size (Å), surface area (m2/g), carbon load (%) and pH stability) of stationary phases that have nominally the same USP column designation, e.g. L1 (see Table 1).
Table 1. Comparison of USP L1 Stationary Phases
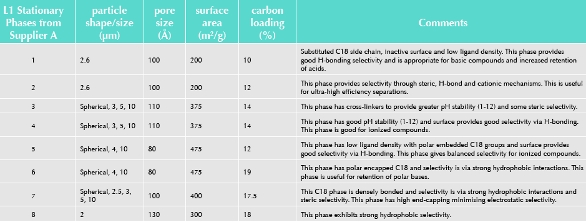
Therefore, to paraphrase Orwell, ‘All stationary phases are equal, but some are more equal than others!’ [18].
The USP and others have tried to address the issue of technology transfer of analytical methods. The USP has introduced a new general chapter, <1224> on this topic [19]. Analytical technology transfer is defined by USP as, ‘the documented process that qualifies a laboratory (receiving unit) to use an analytical test procedure that originates in another laboratory (transferring unit), that ensures that the receiving unit has the procedural knowledge and ability to perform the transferred analytical procedure as intended’ [19]. Technology transfer can be verified by several different options; which include comparative testing, co-validation of methods between laboratories, complete (or partial) re-validation by the receiving unit and a waiver of transfer procedures (typically utilized for pharmacopeial procedures). However, these are very resource intensive approaches to the problem and don’t necessarily address the need to develop better methods with enhanced intrinsic robustness and ruggedness built-into the design phase.
QbD Approaches to Method Development and Transfer
Many companies have independently investigated the intrinsic ruggedness of the myriad stationary phases that are commercially available, with a view to constraining the choice of stationary phases to a restricted number of phases with the knowledge that these phases will produce reliable, fit for purpose methods. Only in those cases where critical resolution(s) cannot be achieved by the default columns would other stationary phases be investigated. Li et al. describe a systematic method development strategy that utilizes six well characterized/ established stationary phases with a diverse range of selectivities [20].
In the recent past, several high-quality publications on the application of QbD principles to analytical measurements [21-23] and HPLC method development [24-27] have been presented in the public domain. A QbD approach can be summarized into four sub-categories: i). Method intent (clear definition of key objectives), ii). Method design (selection of method parameters), iii). Method evaluation (assessment of prototype method) and iv). Method control (implementation of a control strategy). Nethercote et al. have also likened method validation to process validation and described the method lifecycle similarly using three subcategories: Stage 1-Method Design, Stage 2-Method Qualification, Stage 3-Continued Method Verification [28].
There is now an increasing willingness to design reliability/quality into analytical methods by first intent, using multi-factorial robustness assessments and ruggedness studies during method development (within the ‘method design’ phase of an analytical method’s lifecycle) [21]. Thus, by designing the intrinsic operating conditions as a design space (known as a method operable design region or MODR)) with known tolerances rather than discrete points, the possibility of method failure is minimized and the method flexibility is increased and the method is intrinsically more robust [26, 27]. A prerequisite for HPLC robustness is a pre-defined critical resolution factor which can be defined a threshold value. A number of different groups have used resolution plots, which relate critical method parameters to critical resolutions to facilitate the definition of robustness for the method [24-25]. To facilitate further method reliability and flexibility Pohl et al. have proposed the use of the ATP [12]. The ATP can be used to describe the analytical method requirements needed to adequately measure the defined critical quality attributes (CQAs) of the drug substance or product. It is then the ATP which would drive the design and development of appropriate analytical methods.
References
- Commission Direction 2003/94/EC, Guidelines on good manufacturing practice in respect of medicinal products for human use and investigational products for human use, 08th October 2003. http://ec.europa.eu/health/files/eudralex/vol-1/dir_2003_94/ dir_2003_94_en.pdf Accessed on 31st May 2013.
- Pharmaceutical Quality Systems, ICH Q10. EMA/INS/ GMP/79818/2011. 31st January 2011.http://www.ema.europa. eu/docs/en_GB/document_library/Scientific_guideline/2009/09/ WC500002871.pdf Accessed on 31st May 2013.
- Concept paper on the revision of chapter 6 of the EU GMP Guide Quality Control, 12th October 2010, EMA/INS/ GMP/632654/2010. http://www.ema.europa.eu/docs/ en_GB/document_library/Regulatory_and_procedural_ guideline/2010/11/WC500099260.pdf Accessed on 31st May 2013.
- Pharmaceutical Development, ICH Q8. EMEA/ CHMP/167068/2004 – ICH, June 2009. http://www.ema.europa. eu/docs/en_GB/document_library/Scientific_guideline/2009/09/ WC500002872.pdf Accessed on 31st May 2013.
- M. Swartz, I. Krull. Analytical Method Transfer, LC-GC, Nov 2006
- ISPE Good Practice Guide: Technology Transfer, ISPE, Tampa, Florida, 2003
- PhRMA Analytical Research and Development Workshop, Wilmington, Deleware, 20 Sept 2000.
- S. Scypinski, D. Roberts, M. Oates, J. Etse, Analytical Method Transfer, Pharm. Tech., 84(3), 2002, 84-88
- Validation of analytical procedures: Text and methodology. CPMP/ICH/381/95. June 1995. http://www.ema.europa.eu/ docs/en_GB/document_library/Scientific_guideline/2009/09/ WC500002662.pdf Accessed on 31st May 2013.
- USP General Chapter <1225> Validation of Compendial Procedures. USP 36 http://www.uspnf.com/uspnf/pub/index?us p=36&nf=31&s=0&officialOn=May 1, 2013 Accessed on 03rd June 2013.
- P. J. Borman, M. J. Chatfield, I. Damjanov, P. Jackson. Method ruggedness studies using a risk based approach: A tutorial, Analytica Chimica Acta, 703(2), 2011, 101–113
- M.Pohl, M.Schweitzer, G.Hansen, M. Hanna-Brown, P. Borman, K.Smith, J.Larew, P. Nethercote, . Implications and opportunities of applying the principles of QbD to analytical measurements,. Pharm. Technol. Eur., 22(2), 2010, 29-36
- USP General Chapter <621> Chromatography. USP 34. https://mc.usp.org/sites/default/files/documents/GeneralChapterPDFs/621Chromatography.pdf Accessed on 31st May 2013.
- J. W. Nolan. LC Troubleshooting. System Suitability. http:// www.chromatographyonline.com/lcgc/data/articlestandard//lcgceurope/242004/98295/article.pdf Accessed on 31st May 2013.
- P. J. Borman , M. J. Chatfield , I. Damjanov and P. Jackson. Design and Analysis of Method Equivalence Studies, Anal. Chem., 81 (24), 2009, 9849–9857
- C.Agut, A.Caron, C.Giordano, D.Hoffman, A.Ségalini. Transfer of analytical procedures: A panel of strategies selected for risk management, with emphasis on the integrated equivalence based comparative testing approach. J. Pharm. Biomed. Anal., 56, 2011, 293-303.
- K. Okusa, H.Tanaka, M.Ohira. Development of a new cyano bonded column for high performance liquid chromatography. J. Chromatographr. A., 869, 2000, 143-149.
- Orwell, George. Animal Farm;. New York: Harcourt, Brace, 1954. Print.
- USP General Chapter <1224> Transfer of analytical methods. USP 36. http://www. uspnf.com/uspnf/pub/index?usp=36&nf=3 1&s=0&officialOn=May 1, 2013 Accessed on 31st May 2013.
- Y. Li, G. J. Terfloth, A. S. Kord, A Systematic Approach to RP-HPLC Method Development in a Pharmaceutical QbD Environment, American Pharmaceutical Review, 12(4), 2009, 87-95.
- K.Monks, I.Molnár, H-J.Reiger, B.Bogáti, E.Szabó. Quality by Design: Multidimensional exploration of the design space in high performance liquid chromatography method development for better robustness before validation. J. Chromatographr. A., 1232, 2012, 218-230.
- P. Borman, M. Chatfield, P. Nethercote, D. Thompson, K. Truman, K. The application of quality by design to analytical methods, Pharmaceutical Technology, 31(12), 2007, 142-152.
- F.G.Vogt, A. S.Kord.Development of Quality-By-Design analytical methods. J.Pharm. Sci., 100, 2011, 797.
- I.Molnár. Searching for robust HPLC methods – Csaba Horvarth and the Solvophobic theory. Chromatographia, 62 (supplement), 2005, 7
- K.Jayaraman, A.J.Alexander, Y.Hu, F.P.Tomasella. A stepwise strategy employing automated screening and DryLab modeling for the development of robust methods for challenging high performance liquid chromatography separation: A case study. Anal. Chim. Acta., 696, 2011, 116-124.
- P. Borman, J. Roberts, C. Jones, M. Hanna- Brown, R. Szucs, S. Bale. The Development Phase of an LC Method Using QbD Principles. Sep. Sci., 2, 2010, 2-8.
- M. Hanna-Brown, P.Borman, S.Bale, R.Szucs, , J.Roberts, C.Jones. Development of Chromatographic Methods Using QbD Principles. Sep. Sci., 2, 2010, 12-20.
- P.Nethercote, P. Borman, T. Bennett, G. Martin, and P. McGregor. QbD for Better Method Validation and Transfer, 2010. http://www.pharmamanufacturing.com/ articles/2010/060.html Accessed on 03rd June 2013.
Phil Borman is a manager at GSK within Product Development, at Stevenage, UK. He is a chartered chemist with more than 16 years experience within the pharmaceutical industry. Phil first studied at UMIST University (Manchester, UK), where he obtained his MSc in Chemistry and more recently obtained an MSC in Industrial Data Modelling from De Montfort University (Leicester, UK). Phil has sat on various PhRMA/EFPIA task groups focussed on developing Analytical QbD strategy across the pharmaceutical industry. He has authored 28 publications and given numerous external presentations at scientific symposia on topics such as QbD applied to analytical methods and specification setting.
David Elder is a director at GSK within Scinovo (focused on externalization) at Ware, UK. He is a chartered chemist with nearly 36 years experience within the pharmaceutical industry. David studied chemistry at Newcastle upon Tyne (BSc, MSC), before moving to Edinburgh to study for a Ph.D. in Crystallography. He has 7 patents to his name. Dr. Elder is a member of the British Pharmacopoeia (Expert Advisory Group PCY: Pharmacy), a member of the Analytical Division Council, Royal Society of Chemistry (RSC), UK and a council member of the Joint Pharmaceutical Analysis Group, UK. He is a member of the Chartered Quality Institute, and a fellow of the RSC. He has authored 59 publications and given 85 external presentations at scientific symposia including several on rationale method development. He has authored ten book chapters and coedited one book (Analytical Characterisation and Separation of Oligonucleotides and their Impurities).