Introduction
20th century English journalist Malcolm Muggeridge once said, “All new news is old news happening to new people.” This quote seems oddly appropriate when considering the recent interest in continuous processing within the pharmaceutical industry. Continuous processing technologies have revolutionized the auto, food, electronics and commodity chemical industries just to name a few. Why then has the pharmaceutical industry resisted continuous processing in favor of traditional batch infrastructure for so many years? That answer is tied to several factors including the large capital investment needed in manufacturing plants, the highly uncertain nature of the product pipeline, relatively low volumes, short product lifecycles, high margins, complex regulatory approval process, solids handling, and the desire to run a wide range of chemical processes in a single equipment set. Over the past ten years, the climate has changed and Pharma has experienced increased pressure to better serve patients through lowering costs, improving efficiency and employing new approaches to improve product quality (QbD). It is no surprise that many of these challenges can be met though the implementation of continuous processing approaches which helps explain the surge in recent interest. An exciting aspect to this more recent work in continuous is that it involves the blending of so many different disciplines such as chemists (synthetic, bio & analytical), engineers (¬chemical, mechanical & automation), as well as modeling experts. Additionally we have seen growth in the number of collaborations between academic laboratories and Pharma centered around continuous. With such a collaborative approach, the field has grown significantly and Pharma is playing a significant role as continuous technologies are integrated in the synthesis of new drug candidates. Since 2006, the product development group at Eli Lilly and Company has been committed to developing continuous processing capabilities from lab scale to manufacturing. These efforts have led to increased speed to delivery for early phase assets and more recently integration into manufacturing. This article will focus on several case studies that illustrate key drivers for why one might choose continuous processing over batch in the synthesis of drug substance.
Reaction Types & Platforms
The process of deciding when and where to apply a continuous processing unit operation is not always straightforward. There may be strong drivers to employ continuous based upon the chemistry demand. Alternatively continuous may also impact business drivers such as capital investment avoidance, environmental impact and new approaches to regulatory filing (QbD).
Chemistry drivers may include:
- Fast reaction kinetics (case 4)
- Energetic reactions (case 4)
- Unstable intermediates or products (case 3, 4)
- High pressure reactions (case 1, 3)
- Temperature extremes (case 2, 3)
- Selectivity (case 3)
Business drivers may include:
- High or low peak demand (case 5)
- High containment (case 5)
- Hazardous materials (case 1)
- Environmental (case 1, 4)
- Quality (case 1,3,4)
- Reduced capital cost (case 1,5)
- Speed to delivery (case 1,2,3)
In 2010, an analysis of the Lilly development chemistry portfolio was done from very early candidate stage (mostly discovery synthesis) to mature manufacturing routes. Reactions were classifi ed across all stages of development and special attention was given to which types of chemistry represented opportunity for continuous. Additionally we examined what new reaction types were emerging from the early phases of development (metal catalyzed chemistry) as well as reaction types that were deemed useful but underrepresented (aerobic chemistry for example). Based on this analysis, several chemistry & separation platforms were identifi ed for further development.
Chemistry Platforms:
- Vapor liquid reactions (H2, CO, etc)
- Extreme conditions
- Metallation chemistry
- Catalysis
- Immobilized reagents
- Aerobic chemistry
Separations Platforms:
- Continuous crystallization
- Continuous crystallization with particle engineering (API)
- Multistage counter-current extraction
- Continuous distillation
Two Main Categories of Continuous Reactors
The recent interest in continuous processing has led to an explosion of new commercially available products and one could very easily be overwhelmed by all of the options. Fundamentally the landscape is much simpler since there are just two main categories of continuous reactors, Continuous Stirred Tank Reactors (CSTR) and Plug Flow with Dispersion Reactor (PFDR). If designed to minimize axial dispersion the PFDR model can be simplifi ed to a PFR model representing ideal plug fl ow. Figure 1 shows simplifi ed sketches for the continuous reactor types.
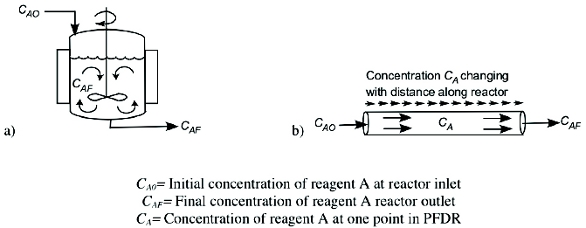 Figure 1. a) Continuous Stirred Tank Reactor (CSTR) and b) Plug Flow with Dispersion Reactor (PFDR)
Figure 1. a) Continuous Stirred Tank Reactor (CSTR) and b) Plug Flow with Dispersion Reactor (PFDR)Reactions performed in CSTRs and PFDRs are each governed mathematically by well-established design equations, as described in chemical engineering textbooks [1-4]. In an ideal CSTR, there is no change in conversion or composition with position inside the reactor. Reagents are diluted by mixing with the bulk contents as soon as they enter the reactor (infi nite dispersion), and composition of the product stream fl owing out is the same as the bulk composition. The PFDR, on the other hand, does have change in conversion and composition with distance along the fl ow path within the reactor from inlet to reactor outlet. PFDRs include micro reactors, static mix tube reactors, open tube reactors, pipe reactors, oscillatory baffl ed tube reactors, spinning tube in tube reactors, and spinning disc reactors. They may also have alternative energy inputs like microwave, photo-, electro-, or sono- energy. Thus, in the PFDR category, there are many diff erent types of reactors, but they share the common feature that they are still governed by the PFDR design equation. Any of the PFDR types of reactors mentioned above can operate in fl ow regimes where dispersion can be neglected if they are designed for minimizing axial dispersion. PFRs and CSTRs off er diff erent advantages and proper evaluation will lead a manufacturer to choose the one that best suits the chemistry. In the following discussion, case studies 1-3 utilized PFDRs, and case studies 4-5 utilized CSTRs because of solid reagents (case 4) and liquid-liquid mixing with long reaction time (case 5).
Case Study 1: Hydroformylation of Methylmethacrylate
Starting material 4 was an intermediate in the synthesis of a late phase drug candidate. The original approach to 4 involved a three step synthesis starting with alcohol 1. Oxidation of alcohol 2 aff orded aldehyde 3 which was then converted to 4 via reductive amination. The TEMPO oxidation used in the second step was problematic and required isolation from high boiling tetralin to avoid product degradation and potential for thermal runaway. Eff orts to scale this chemistry in the CMO network led to very poor results leaving the team without this key intermediate and jeopardizing supply of the API for clinical studies. However, a solution was brought forward in rapid fashion involving the continuous hydroformylation of methylmethacrylate (5) in a pulsating fl ow tube reactor with purifi cation by continuous multi-stage distillation. Over 175 kg of highly pure aldehyde 3 was produced in a hydrogenation bunker and a traditional walk-in laboratory hood in 14 days of operation. The continuous approach to 3 utilized a cheaper starting raw material, required one less step, required no co-solvent, and aff orded 3 in higher yield and purity than the original route. In fact, the process mass intensity (PMI) of the hydroformylation route was just 1.75 compared to a PMI of 128 for the TEMPO route [6]. Hydroformylation is an example of a reaction type with hazardous reagents, H2 and CO, which can be run more safely continuous rather than batch. Safety issues around the use of H2 and CO were mitigated by using a smaller continuous reactor (compared to batch) that operates almost completely liquid fi lled. This results in reduced volume of pressurized hazardous gas which is inherently safer than batch approaches.
 Scheme 1: Batch & Continuous Approaches to API SM 4
Scheme 1: Batch & Continuous Approaches to API SM 4Case Study 2: Thermal Rearrangement
One of the most utilized continuous platforms is a simple thermal tube where reagents and solvents can be rapidly heated to high temperature, often well above the boiling point of the solvent. At research scale, these reactions are often conducted in sealed tube systems with microwave heating, but this is not practical at larger scale. One example of a thermal rearrangement is the Ortho-Claisen (OCR) thermal rearrangement which requires temperatures above 200 °C.
The optimization and scale up of and Ortho-Claisen rearrangement (7 → 8) was performed safely using a continuous reactor operating at 230°C and 4 hour τ (Scheme 2) [7]. The ideal operating ranges for yield, selectivity, and throughput were outside of those allowed for straightforward batch processing. In the fl ow reaction at 230 °C, the temperature was easily achieved in a thermal tube, temperature profi le was scaled with little variation, and the reaction ran more concentrated while mitigating the safety hazards. The extremely high surface area per unit volume (A/V) for heat transfer in tubular fl ow reactors allows for rapid and scalable heating and cooling. For example, at comparable throughput rates, the A/V was 2050 m2/m3 for the fl ow tube reactor, compared to 42 m2/m3 for a batch reactor. This was a fast, safe alternative to batch for scale up to early phase material delivery.
 Scheme 2: Ortho Claisen Rearrangement of Allylether 7
Scheme 2: Ortho Claisen Rearrangement of Allylether 7Case Study 3: Thermal Cyclization
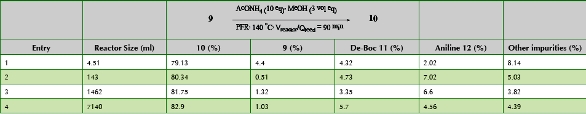
One of the main diff erences between running in a PFDR versus a traditional batch tank is the ability to “scale time”. The high surface area to volume ratio in a PFDR allows for rapid heating on the reactor inlet and cooling at the outlet. In a batch tank however, as a scientist increases scale, the amount of time to heat and cool will be greater. One of the trends a process chemist looks for during development is any scale dependent reaction performance issues as this may indicate that long heating cycles could result in poor performance. Such was the case for the cyclization of ketoamide 9 (Scheme 3) [8]. Research scale batch cyclizations of 9 were conducted in n-butanol at refl ux and aff orded a mixture of desired imidazole 10, deprotected product 11 and animine impurity 12. Increasing the batch reaction size from 1g → 50 g → 200 g resulted in a steady decrease in product formation (-12%) and increase in impurity load (+8%). The origin of the problem was more fully understood when a stability study of 9 was conducted in the presence of ammonium acetate at lower temperatures (Figure 1). Rapid loss of potency was observed at temperatures well below the preferred reaction temperature. Therefore any increased time required to reach a productive temperature regime would result in signifi cant degradation of the starting material.
 Scheme 3: Cyclization of Ketoamide 9 with Ammonium Acetate
Scheme 3: Cyclization of Ketoamide 9 with Ammonium Acetate Figure 2. Stability of 9 at lower temperatures
Figure 2. Stability of 9 at lower temperaturesThis scenario was ideally suited for continuous processing where high A/V allows for very rapid heating of reaction solutions. Table 1 nicely shows reproducible reaction performance over a wide range of PFR sizes. Over 29 kg of 10 was produced using a 7L coiled tube reactor after workup and isolation. Methanol was chosen as reaction solvent because it simplifi ed workup and isolation of 10. High pressure (>160 psig) was needed because desired reaction temperature was far above the boiling point of methanol. This was a fast, higher quality alternative to batch for scale up to early phase material delivery.
Table 1. Summary of Cyclization of 9 Using Different Plug Flow Reactor Sizes and Throughputs
Case Study 4: Continuous Flow Barbier Reaction
 Figure 2. Continuous Barbier Laboratory Setup
Figure 2. Continuous Barbier Laboratory SetupFor 100 years, Grignard reactions have been one of the most powerful and effi cient organic chemistry methodologies for C-C bond formation. However, Grignard reactions are also among the most challenging reactions from both operational and potential safety issues due to initiation diffi culties and runaway potential. A close variation to the Grignard reaction is the Barbier reaction wherein the Grignard reagent is prepared in the presence of an electrophile resulting in the immediate consumption of the Grignard. A Barbier reaction using a CSTR was developed for a key pharmaceutical intermediate in production of edivoxetine·HCl (Scheme 4) [9]. In the fl ow setup (Figure 2), solid magnesium is sequestered in the fi rst tank where the Grignard initiation event takes place. CSTR 2 was used as an aging tank and CSTR 3 was the quench tank. CSTRs were used for Grignard reaction rather than a PFDR because of the solid Mg reagent.
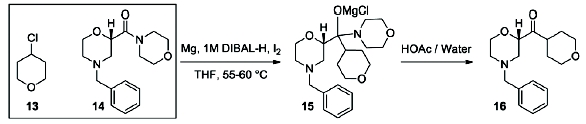 Scheme 4: Barbier Reaction to form Ketone 15
Scheme 4: Barbier Reaction to form Ketone 15Continuous reaction improved process safety, product quality, and process greenness. The continuous reaction achieved >99% ee in situ versus 95% ee batch because of immediate conversion of unstable intermediate. Solvent volumes were reduced 30%. The safety hazards were reduced by decreasing the reactor size by 50X, which reduced chemical potential and also increased heat transfer surface area per unit volume by 4X. DIBAL-H initiating agent was reduced by more than 100X, and excess Mg that must be quenched at the end of reaction was almost eliminated. When run continuously, the commercial scale Grignard formation reactor was expected to be 50L, which replaces 4000-6000L batch reactor.
Case Study 5: Cytotoxic Containment
Continuous processing has long been associated with high volume product, but there are also drivers for low volume products as well. Continuous can enable cytotoxic API production in inexpensive, dedicated, “disposable” equipment sets for production of low volume (< 1000 kg/year) cytotoxic compounds in laboratory fume hoods. This approach was taken for the development and scale-up of a synthetic route to LY573636·Na (Scheme 5) [10]. The fi nal two steps of the synthetic route were telescoped using continuous fl ow Schotten−Baumann conditions which resulted in fewer unit operations than the existing batch process, eliminated an isolation of a cytotoxic intermediate, and ran at commercial scale (5kg/day) using small inexpensive laboratory glassware and fl exible tubing. The reaction, extraction, distillation, and crystallization platform technologies were designed to run fully continuously in standard walk-in lab hoods. This was a low cost alternative to increasing cytotoxic capacity in manufacturing with high containment. CSTRs were used rather than a PFDR for the reaction because of the 2 immiscible liquid phases, the long reaction times, and the laminar fl ow regime. Continuous counter-current multi-stage liquid liquid extraction benefi tted dramatically by the use of continuous rather than batch because of the high water solubility of LY573636·Na. Extraction was the key impurity control point for the sodium salt of benzoic acid, which was purged with high effi ciency while maintaining high yield because of the counter-current operation.
 Scheme 5: Schotten−Baumann Reaction to form LY573636·Na
Scheme 5: Schotten−Baumann Reaction to form LY573636·NaNetwork Partner Needs and Platform Technologies
If a process step will be part of the registered sequence, but if the inhouse manufacturing facility will not run the process step until later phase campaign such as registration stability or validation campaign, then a network partner is needed for outsourcing the process for GMP production in earlier phases of development. Furthermore, if the process step will be in the pre-registered sequence, or an earlier registered step that will remain outsourced to the network after launch, then a network partner is needed for outsourcing the process through commercialization. This means that outsourcing partners must be capable of running the continuous reactions and continuous separations unit operations. Often the network partner will also have research scale experimental capabilities for the continuous reactors and separators. Based on the examples given in this paper, Table 2 provides a summary of platform technologies for continuous processing, along with typical equipment size and volumetric throughput for research, pilot, and commercial scale manufacturing.
Table 2. Platform Technologies for Continuous Processing
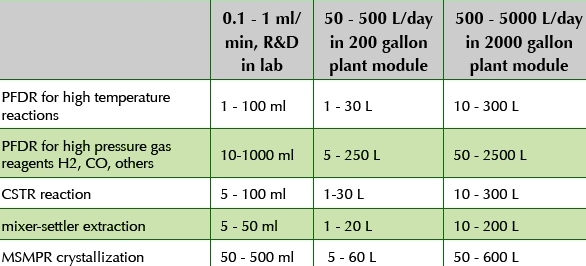
A goal is to keep the continuous reaction, extraction, and crystallization skids portable, flexible, simple, and versatile. The pumping skids are generally kept separate from the reaction or separations unit operation skids, so that the same pumping skids can be used for a wide variety of continuous processes.
Mixer-settlers are appropriate for extraction in the pharmaceutical industry for several reasons. This extraction technology tolerates wide flow rate ratio of two liquid phase from 1:1 up to greater than 20:1, small density ratio for example 1.02:1, operate in cross-flow mode for all stages, counter-current for all stages, or any combination, startup and shutdown the process at the beginning and end of a campaign with no transition waste, stop and restart process easily at any time in the middle of the run with no transition waste, and permit any number of continuously flowing feed streams to any stage.
Compared to batch crystallization, continuous crystallization can offer several advantages. In some processes continuous crystallization can result in better impurity rejection, prevent oiling, use wider volume ratios solvent to antisolvent, give steady state control of crystal size distribution, increase yield using steady state recycle, and minimize equipment size, cost and cleaning for cytotoxics. Furthermore, continuous crystallization is required for integration to otherwise fully continuous wet end process used to increase throughput.
As shown in the table, equipment size is greatly reduced compared to batch for the same mass per time throughput. However, large feed tanks and product tanks are needed to run the process. For example, if a 100 L continuous reactor with 2000 L/day throughput is used for a high temperature reaction in a plant module with 2000 gallon tanks, and if the workup and isolation is intended to be accomplished batch, then the 2000-gallon vessels may be used as feed tanks and product tanks. Also, inexpensive and portable parallel 1000L surge vessels may be used for feed and product tanks so that the main vessels can be used for the batch unit operations while the feed and product collection vessels are filling and emptying.
Conclusions
This paper provided several case studies that illustrated key drivers for why one might choose continuous processing over batch. There are fundamentally two main categories of continuous reactors, CSTR and PFDR, and both were utilized in the case studies. A high pressure hydroformylation reaction benefitted from continuous versus batch because of safety reasons, and the product isolation was made more efficient with less solvent waste by continuous multistage fractional distillation. Waste was reduced by about two orders of magnitude compared to the batch process. The scale up of an Ortho-Claisen rearrangement was done safely using a continuous reactor operating at 230 °C and 4 hour τ. Heat transfer surface area per unit volume was about 50X higher in a tubular flow reactor compared to batch,which allows for rapid and scalable heating and cooling and reduces the risk of thermal runaway. An imidazole cyclization reaction required fast and scalable heating and cooling times, and precisely controlled time at 140°C, to achieve high selectivity and yield. This reaction was much more scalable continuous than batch. A Grignard formation, coupling, and quench were done in continuous stirred tank reactors. Compared to batch, continuous reaction improved process safety by reducing reactor size 50X, improved product quality by achieving higher %ee, reduced solvent waste, and almost eliminated the excess Mg that must be quenched at the end of reaction. Finally, continuous reaction, extraction, distillation, and crystallization enabled cytotoxic API production in an inexpensive, dedicated, “disposable” equipment set for commercial scale production in laboratory fume hoods. Outsourcing partners must be capable of running the continuous reactions and continuous separations unit operations. Equipment size is greatly reduced compared to batch for the same mass per time throughput. However, large feed tanks and product tanks are needed to run the process.
References
- Levelspiel, O. The Chemical Reactor Omnibook, OSU Book Stores, Inc., Corvallis, Oregon, 1993.
- Levenspiel, O. Chemical Reaction Engineering. An Introduction to the Design of Chemical Reactors; John Wiley and Sons, Inc.: New York, NY, 1962.
- Fogler, H. S. Elements of Chemical Reaction Engineering; Third ed.; Prentice Hall PTR: upper Saddle River, New Jersey, 1999.
- Process Dynamics in Environmental Systems; Walter J. Weber, J.; DiGiano, F. A., Eds.; John Wiley and Sons, Inc. : New York, NY, 1996.
- Braden, Timothy Michael; Gonzalez, Miguel Angel; Jines, Amy Rebecca; Johnson, Martin David; Sun, Wei-Ming. Patent Application WO 2009/023515 A2.
- 2011 President’s Green Chemistry Award Nomination: “Use of Continuous Processing to Enable a Convergent Route to LY2624803*H3PO4”. http://www.epa.gov/greenchemistry/pubs/docs/award_entries_and_recipients2011.pdf
- Rincón, Juan, and Mario Barberis, María González-Esguevillas, Martin D. Johnson, Jeffry K. Niemeier, and Wei-Ming Sun Org. Process Res. Dev., 2011, 15 (6), 1428–1432.
- May, S. A.; Johnson, M. D.; Braden, T. M.; Calvin, J. R.; Haeberle, B. D.; Jines, A. R.; Miller, R. D.; Plocharczyk, E. F.; Rener, G. A.; Richey, R. N.; Schmid, C. R.; Vaid, R. K.; Yu, H. Org. Process Res. Dev., 2012, 16 (5), pp 982–1002.
- Kopach, M. E.; Roberts, D. J.; Johnson, M. D.; McClary Groh, J.; Adler, J. J.; Schafer, J. P.; Kobierski, M. E.; Trankle, W. G., Green Chem., 2012, 14, 1524-1536.
- White, T. D.; Berglund, K. D.; Groh, J. M.; Johnson, M. D.; Miller, R. D.; Yates, M. H., Org. Process Res. Dev., 2012, 16 (5), 939–957
Author Biographies
Scott May received his Ph.D. in organic chemistry in 1998 and joined the process research group at Eli Lilly that same year. In his current role as Senior Research Advisor his focus is on the identification, development and implementation of new innovative technologies to accelerate the drug commercialization process.
Dr. Martin Johnson received his dual doctorate in chemical and environmental engineering in 2000 and then joined Dow Chemical. In 2005 he joined Eli Lilly, where he leads a group of engineers in the continuous processing technology function of R&D, developing platform technologies for continuous reaction and separation unit operations.