Introduction
Among the challenges involved in developing acceptable commercial dosage forms for active pharmaceutical ingredients (API’s) is demonstrating adequate chemical stability [1,2]. Expiration dating is generally determined based on the time a drug product remains within specification limits of potency or of individual or total degradation products. This review introduces a procedure for stability assessments of solid drug products and solid API’s that provides credible predictions for product expiration dating, improves product understanding, reduces uncertainty, and has the potential to change the way the pharmaceutical industry meets its stability commitments for clinical investigations, drug product registration and post-approval changes. This process also significantly reduces the time needed to make stability assessments without adding any risks to patients.
We have called this approach the "Accelerated Stability Assessment Program" (ASAP). This approach consists of four elements which will be discussed below: (1) the concept of isoconversion to compensate for the complexity of solid-state degradation reaction kinetics, (2) a moisture-corrected Arrhenius equation that explicitly takes into account the effect of relative humidity (RH) on degradation rates in the solid state, (3) statistical design and analysis to both provide a reasonable estimation of parameters and explicitly determine error bars for extrapolated shelf-lives, and (4) by combining the effect of RH on stability with the protection afforded by packaging to assess the shelf-life at different storage conditions and packaging configurations.
Isoconversion
In solution, molecules can react to give product with a kinetic rate that is characterized by the order of the reaction; i.e., reaction rates will depend on the molecule’s concentration to a power dependent on the mechanism. In the solid-state, this is complicated by the fact that with low mobility, molecules can exist in multiple, non-equilibrating states. Examples of these states are crystalline bulk, crystalline surface, amorphous material, and material dissolved in other materials (solid solution). Each of these states can potentially react to form product with its own kinetics. The overall kinetics for this heterogeneous situation can become extremely complicated. Even in the simple case of two states, one more reactive, and the other relatively stable, it can be difficult to deconvolute the individual contributions from a product profile. Moreover, one would need to collect multiple time points and carry out a stability experiment to significant drug degradation levels to do so. The concept of isoconversion [3,4] provides an alternate approach. For this concept, one considers only the time required for a degradation reaction to reach the specification limits for loss of potency or formation of degradant(s). While this does not, strictly speaking, provide a chemical reaction rate constant, it still provides a usable rate constant (i.e., specification limit divided by time).
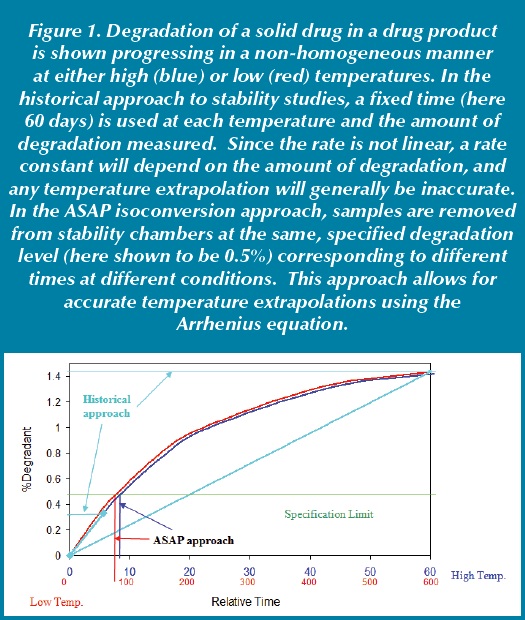
A key attribute of heterogeneous kinetics in solid drug products is that while rates change with temperature, the curve shape of the degradant (or loss of API) versus time plot remains more or less constant. This is true because generally the differences in reaction rates of the different states are mostly due to mobility rather than factors likely to influence activation energies. Because of this, employing the isoconversion concept provides a new way of conducting accelerated stability studies which can increase accuracy. To understand this, consider the historical approach for accelerated aging and stability studies in general: Protocols traditionally used are generic with fixed time points at different storage conditions. The amount of degradant formed (or potency lost) is determined at these time conditions which is then used to estimate the behavior at an extrapolated condition with respect to time and temperature. In contrast, the ASAP isoconversion approach works somewhat in the opposite mode. Instead of keeping time points fixed and determining the amount of degradant/potency change, isoconversion keeps the amount of degradant/potency change constant at the specification limit and varies the time. Since with most drug products (about 65% in our experience), formation of degradant (loss of potency) is not linear with time, the rate measured for the historical approach will vary with the extent of conversion: typically rates slow as more reactive API states are consumed. The result is that the effective rate constant at any storage condition and time will not be reflective of the rate to a different conversion level since varying amounts of each API state is involved. In contrast, with the ASAP isoconversion approach, the rate constant at every storage condition will have the same relative contributions from the different solid-state environments. The difference between the two approaches is illustrated in Figure 1. While the historical approach allows generic protocols across a company’s product portfolio, in many cases it provides inaccurate predictions especially under high temperature conditions. In fact, many cases where drug products are considered to have anomalous behavior at high temperatures can become well-behaved if the isoconversion approach is used. It should be noted, however, that high temperature studies may not be predictive of ambient stability if there is a change in API or excipient physical form (e.g., a melting point) with the temperature range being used.
Relative Humidity Effects on Solid-state Drug Degradation
Solid API and drug product chemical stability is affected by the RH that the sample experiences. The moisture-modified Arrhenius equation (Equation 1) quantifies drug product stability as a function of temperature and RH [1,2]:

where k is the degradation rate (typically percent degradant generated per day), A is the Arrhenius collision frequency, E a is the energy of activation for the chemical reaction, R is the gas constant (1.986 cal/(mol K)), T is the temperature in Kelvin, and B is a humidity sensitivity constant which has been found to vary from 0 to 0.10. The form of Eq. 1 indicates that chemical instability increases exponentially with an increase in RH. RH can have a very significant effect on chemical stability, depending on the B-term. For example, with B equal to 0.09, a shelf-life at 60%RH of 5.0 years would drop to only 1.2 years at 75%RH (without packaging protection). Because of the significance of the RH, accelerated stability studies that do not explicitly control RH will not be predictive. In fact, if packaged product is used for accelerated stability studies, the RH the sample is actually exposed to must be accounted for to provide accurate predictions even if the external RH is controlled. Since many historical accelerated stability studies do not compensate or control properly for RH, the data are often erratic and non-predictive.
Statistical Design and Analysis
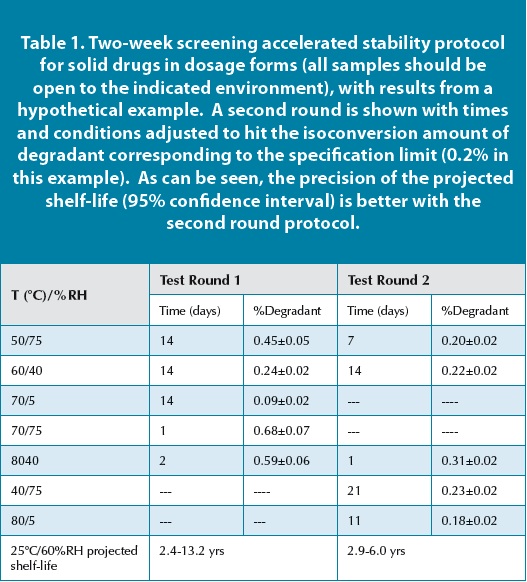
Equation 1 provides the framework for the explicit determination of the isoconversion rate constant at any condition of temperature and RH. In order to take advantage of this capability, three terms must be determined from a data set: A, Eaand B. Fitting these parameters requires a minimum of three experiments where temperature and RH are varied. In practice, it is generally advisable to use designs for varying temperature and RH that involve at least five experiments (two degrees of freedom). Ideally, these experiments will involve storage of samples (API or drug product) at times that provide for degradation corresponding to isoconversion. In addition, it is important that the samples be stored at the RH under open conditions so that the RH of the samples is at a known equilibrium value. An example of a screening protocol that provides reasonable data for most solid dosage forms is shown in Table 1. API’s tend to be more stable, so this protocol may be too mild to hit specification limits for many API’s. Since this protocol assumes one knows the degradant level before actually having measured values, determining the appropriate conditions for a given sample can require multiple iterations. Fortunately, in the drug development process, early assessments of stability can have a wider range of uncertainty. As product development proceeds through different phases, protocols become more refined, and greater precision (and accuracy) is provided for the corresponding ambient shelf-life. This is shown in Table 1 in the form of an example where the screening protocol was refined based on the data to give a more precise stability determination in the second round.
Once data are generated, statistical analysis is important to propagate errors from measurements through to projected conditions. We have found that use of a Monte-Carlo simulation process provides a method for the estimation of shelf-life (use period). A number of consequences fall out of the statistics: (1) The precision of the analytical method will have a direct effect on the precision of the final shelf-life prediction; (2) The greater the extrapolation distance on the temperature or RH axis, the greater the error bars in the predictions. This means that highly accelerated stability studies (e.g., high temperature, high RH) may provide quite accurate predictions of ambient stability in very short times, but the confidence interval can be large. For some drug products and API’s, even a large range can still provide an acceptable determination. For others, typically when estimations fall between one and four years, greater precision may be needed. This precision can typically be achieved by some combination of adjustments to extrapolation distance (e.g., lower temperatures with longer storage times) or by increasing the sampling at each condition.
Packaging
For drug products, packaging can provide protection from the external RH. With ASAP, one can determine the effect of RH on drug degradation rates. Below, a method for calculating the RH as a function of time in packaging is shown. By combining the ASAP-determined degradation rate as a function of RH with the determination of the RH as a function of time inside packaging, it becomes possible to explicitly determine the amount of degradant formed as a function of time for a given packaging at a given storage condition [5-8].
Determining the effect of packaging on the RH inside the package over time involves two key elements: moisture transfer into or out of the package and equilibration of moisture within the package. Equilibration of moisture inside a package is so much faster than moisture transfer into or out of a package that one can assume that the internal moisture of the pharmaceutical materials and the headspace inside a package is always at equilibrium.
Moisture transfer into or out of a package can be described in terms of a moisture vapor transmission rate, MVTR. The MVTR of a package (bottle, blister) is the mass (in units of mg H2O/day) of water permeating into the headspace volume for a given temperature and difference between internal and external RH. When an empty container (i.e., one with no components or moisture) is placed in a stability chamber, moisture from the chamber environment will permeate through the walls into the headspace at a rate related to the MVTR and the difference in RH. With no water adsorbing components, any moisture transferred must directly accumulate into the bottle headspace, changing the RH accordingly. As an example, with a 60-cc, high-density polyethylene (HDPE) sealed pharmaceutical bottle, the internal RH goes from 20% to 75% RH in only about ten days when stored at 40°C/75%RH.
Moisture inside a package will equilibrate among the components (i.e., tablets, powders, capsules and desiccants) based on how much moisture each component holds at a given RH. Materials will adsorb moisture to different degrees as described by their moisture sorption isotherms, which is a plot of water percent (w:w) in the material as a function of the RH. The moisture sorption behavior of most materials can be described according to the Guggenheim-Anderson-de Boer (GAB) equation [9-11]. Because the moisture sorption isotherm of a combination of multiple components can be modeled using the weighted sum of the GAB parameters for those components, one can calculate the moisture sorption isotherm for a drug product based on the summation of the GAB parameters of the combined formulation components. Similarly, desiccants are materials that adsorb moisture with a relatively high capacity and thereby are effective at keeping the RH inside a package lower than it would otherwise be, though it will still increase with time.
The RH as a function of time inside a package can therefore be calculated quite accurately by combining information about the initial RH of the internal components, their corresponding moisture sorption isotherms and the MVTR of the package. This RH dependence can be coupled with the explicit knowledge of the RH dependence of the API reactivity according to Eq. 1 to give a point-by-point determination of degradation as a function of time. The result is that the degradation level for solid API’s and drug products in packaging can be determined accurately even using accelerated stability studies. An example of the RH and degradant level calculations and experimental validation is shown in Figure 2.
Conclusion
An isoconversion paradigm, where times in different temperature and humidity-controlled stability chambers are set to provide a fixed degradant level at the specification limit, is introduced to compensate for the heterogeneity of solid drugs and drug products. Reliable estimates for temperature and RH effects are handled using a humidity-corrected Arrhenius equation. A statistical protocol is employed to determine best fits for the stability data, which in turn allows for precise estimations of shelf-life at any storage condition. Once the RH dependence of a drug or drug product’s stability is determined, the shelf-life inside packaging can be calculated knowing the MVTR and moisture sorption isotherms of the internal components. These methodologies provide far better predictions of shelf-life (expiry) than previously possible in a significantly reduced time frame. Precise shelf-life estimations can be made using a two-week, product-specific protocol, which allows for formulation development, package selection, expiry setting and determination of the effects of post-approval changes on process or materials [12]. This review shows how the science of solid-state reaction kinetics can be used to more effectively speed the drug development process.
References
1. "Accelerated aging: prediction of chemical stability of pharmaceuticals", Kenneth C. Waterman and Roger C. Adami, International Journal of Pharmaceutics (2005), 293, 101-125.
2. "Stress testing: a predictive tool", Steven W. Baertschi and Patrick J. Jansen, Drugs and the Pharmaceutical Sciences (2005), 153 (Pharmaceutical Stress Testing), 13-49.
3. "Improved protocol and data analysis for accelerated shelf-life estimation of solid dosage forms", Kenneth C. Waterman, Anthony J. Carella, Michael J. Gumkowski, Patrick Lukulay, Bruce C. MacDonald, Michael C. Roy and Sheri L. Shamblin, Pharmaceutical Research (2007), 24, 780-790.
4. "Understanding and predicting pharmaceutical product shelf-life", Kenneth C. Waterman, In Handbook of Stability Testing in Pharmaceutical Development: Regulations, Methodologies and Best Practices, Kim Huynhba, Ed. Springer Science and Media Publishing, Chapter 6 (2008) 115-135.
5. "Package selection for moisture protection for solid, oral drug products", Kenneth C. Waterman and Bruce C. MacDonald; manuscript in preparation.
6. "Predicting drug hydrolysis based on moisture uptake in various packaging designs", Klemen Naveršnik and Simona Bohanec, European Journal of Pharmaceutical Sciences (2008), 35, 447-456.
7. "The effects of packaging on the stability of a moisture sensitive compound", Jennifer G. Allinson, Richard J. Dansereau and Adel Sakr, International Journal of Pharmaceutics, (2001), 221, 49-56.
8. "Application of sorption-desorption moisture transfer modeling to the study of chemical stability of a moisture sensitive drug product in different packaging configurations", Sherif I. F. Badawy, Anthony J. Gawronski and Francisco J.Alvarez, International Journal of Pharmaceutics, (2001), 223, 1-13.
9. "Evaluation of the moisture sorption behavior of several excipients by BET, GAB and microcalorimetric approaches", Robert Roskar and Vojko Kmetec, Chemical and Pharmaceutical Bulletin, (2005), 53, 662-665.
10. "Processing and storage effects on water vapor sorption by some model pharmaceutical solid dosage formulations", Chad R. Dalton and Bruno.C. Hancock, International Journal of Pharmaceutics, (1997), 156, 143-151.
11. "A study on moisture isotherms of formulations: the use of polynomial equations to predict the moisture isotherms of tablet products", Yanxia Li, Yeshwant D. Sanzgiri and Yisheng Chen, AAPS PharmSciTech. (2003), 4, 1-8 (article 59).
12. "A science-based approach to setting expiry dating for solid drug products", Kenneth C. Waterman and Stephen T. Colgan, Regulatory Rapporteur, (2008), 5 9-14.
Ken Waterman received his B.S. degree with honors in chemistry from UCLA where he carried out syntheses of asymmetric transition metal catalysts. He received his Ph.D. in physical-organic chemistry at UC Berkeley (with Professor Andrew Streitwieser) conducting work on acidity, computational chemistry and synthesis of highly charged organic molecules, followed by two-years of post-doctoral studies in physical chemistry as an NIH research fellow at Columbia University (with Professor Nicholas Turro) on solid-solid, solid-liquid and solid-gas interfacial chemistry. In 1987, Dr. Waterman began a 12-year career at Polaroid Corp. in Massachusetts where he invented and brought to market a number of new and modified imaging systems, both for photographic and digital applications. Since 1999, he has been in the Pharmaceutical Sciences Division at Pfizer (Connecticut), currently as a Research Fellow. His research interests include drug stability, drug delivery technology and biopharmaceutics. He is the author of over 60 journal articles, book chapters and US patents.