Introduction and Background
A significant body of scientific and regulatory knowledge has been accumulated over the past two decades regarding “leachables” in various drug product types and dosage forms. Leachables are chemical entities, either inorganic or low molecular weight organic, that migrate (i.e., leach) into a drug product over the shelf-life of that particular drug product. The majority of leachables that have been observed originate from various components of the drug product container closure and packaging systems. These components (e.g., bottle caps, labels, seals, etc.) can be fabricated from materials such as rubber and plastic which usually contain functional chemical additives and other chemical entities that can be potential leachables. The functional additives are typically low molecular weight (i.e.,<1500 amu) organic compounds with a wide variety of chemical structures, and by definition perform various functions [1] within the component materials of construction; such as antioxidants, UV-stabilizers, polymerization initiators and accelerators, anti-static agents, pigments, antislip agents, plasticizers, lubricants, etc. Also included within the broader category of potential leachables are:
- Monomers and oligomers resulting from incomplete polymerization processes in elastomeric and plastic materials (e.g., dimers, trimers, tetramers, etc. from polybutyleneterephthalate; PBT).
- Surface residues and other chemical entities from component fabrication processes and other drug substance/drug product manufacturing processes (e.g., drawing oil residues on metered dose inhaler metal canister surfaces).
- Chemical entities contained in materials external to, but in direct contact with, the primary drug product container closure system (e.g., vanillin volatilized from cardboard shipping containers).
- Degradation products from, and trace level impurities present in, the primary functional additives to rubber and plastic components (e.g., N-nitrosamines present at ppb levels in certain sulfur-cured rubbers).
Table 1 - FDA/CDER/CBER risk-based approach to consideration of leachables
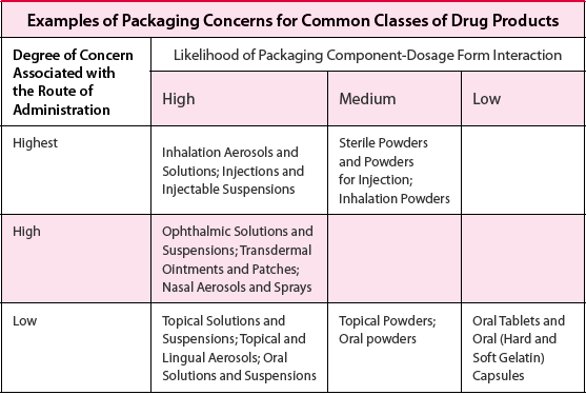
The USFDA Centers for Drug Evaluation and Research (CDER) and Biologics Evaluation and Research (CBER) have stated in their “Guidance for Industry; Container Closure Systems for Packaging Human Drugs and Biologics; Chemistry, Manufacturing and Control” [2] that for qualification and control of packaging components, the type and extent of information that should be provided in a regulatory submission depends on the dosage form and the route of administration. This can be thought of as a risk-based approach relative to, among other considerations, the potential exposure of patients to drug product leachables. Table 1 is adapted from this guidance document and categorizes various dosage form types relative to these two central criteria of risk. Dosage forms which carry the highest risk relative to leachables exposure include various inhalation drug products (also referred to as OINDP; Orally Inhaled and Nasal Drug Products) and certain types of injectable drug products, while those with the lowest risk include various solid oral dosage forms. Based on this risk assessment, inhalation drug products (OINDP) have received the greatest attention from both regulatory authorities and the pharmaceutical industry regarding leachables, and it is easy to see why since the majority of inhalation drug products are intended for chronic administration to the already compromised respiratory system of a sensitive patient population. In fact, two additional USFDA guidance documents exist specifically for these drug product types, including one for Metered Dose Inhalers (MDIs) and Dry Powder Inhalers (DPIs) [3] and one for Nasal Spray and Inhalation Solution, Suspension and Spray Drug Products [4]. The Product Quality Research Institute (PQRI) has sponsored a working group which issued a recommendation document [5], along with additional publications in the scientific literature [6, 7], for development of new inhalation drug products relative to leachables. This PQRI working group developed the first ever safety-based qualification (5 μg/day Total Daily Intake) and assessment (0.15 μg/day Total Daily Intake) thresholds for leachables in any drug product type, although these thresholds are intended to apply only to inhalation drug products. The working group also developed best practice recommendations for leachables characterization, assessment and control in inhalation drug products.
For inhalation and other high-risk dosage forms, it is necessary to understand “potential leachables” in order to adequately detect, identify, qualify, and control actual drug product leachables. Potential leachables can be characterized in two complementary ways: 1) evaluation of the composition of appropriate container closure system components based on information provided by component suppliers and fabricators; and 2) laboratory investigations into the identities and quantities of potential leachables that can be extracted from appropriate container closure system components under relatively vigorous conditions. These laboratory investigations into potential leachables are referred to as Controlled Extraction Studies, and the chemical entities identified and quantified are referred to as “extractables”. It is always true that drug product leachables are a subset of extractables, and can be correlated either directly or indirectly with extractables [5]. For inhalation drug products and other high-risk dosage forms, regulatory submissions for new drug products typically require extensive information related to leachables and extractables, including:
- Complete information on the chemical compositions of all appropriate container closure system and other packaging components (i.e., potential leachables).
- Controlled Extraction Studies for all appropriate container closure system and other packaging components (i.e., potential leachables).
Note that for high-risk dosage forms it can never be assumed that information from component suppliers regarding potential leachables is adequate for a regulatory submission. - Drug product leachables studies under appropriate stability storage conditions.
- Qualitative and quantitative correlations of leachables with extractables.
- Appropriate safety assessment and qualification of observed drug product leachables.
- Extractables and leachables specifications and acceptance criteria based on scientific evaluations.
Along with the safety-based thresholds for leachables assessment and qualification, the PQRI working group’s publications include so-called “best practice” recommendations for container closure system component selection, Controlled Extraction Studies, leachables studies, and routine extractables control [5, 7].
In contrast, new drug product regulatory submissions for low-risk dosage forms such as solid orals do not require this extensive level of information regarding leachables and potential leachables. For example, Controlled Extraction Studies and leachables stability studies are not routinely accomplished for container closure system components and drug products. The information typically submitted for such dosage forms includes [2]:
- Descriptions of materials of construction and any additional treatments to these materials.
- Chemical compositions of all plastics, elastomers, adhesives, etc.
- References to indirect food additive regulations.
- USP Physicochemical and Biological tests (i.e., USP 661 and 381; USP 87/88).
However, low risk does not mean zero risk. For example, Fang, et al. [8] detected unknown drug product impurities above the appropriate ICH threshold [9] in a tablet formulation during an evaluation of new inks and label coatings. LC/UV (Liquid Chromatography/Ultraviolet detection), GC/MS (Gas Chromatography/Mass Spectrometry) and LC/MS (Liquid Chromatography/ Mass Spectrometry) identified these impurities as the photoinitiators: 1-benzoylcyclohexanol and 2-hydroxy-2-methylpropiophenone. Retrospective Controlled Extraction Studies traced these leachables to the ink and label coating on the low-density polyethylene (LDPE) drug product bottles. The photoinitiators had migrated from the ink and label coating through the plastic bottle and into the tablets.
Leachables typically present themselves in solid oral dosage forms and other low-risk drug product types as “unspecified” drug product impurities in an LC/UV (or other type of) impurity profile. Such leachables are identified using processes typically employed for the identification of any other unspecified drug product impurity [10, 11]. Once an unspecified impurity has been identified as a leachable, a process of “reverse-engineering” involving Controlled Extraction Studies on container closure system components, is used to discover the origin of the leachable, as with Fang, et al. [8]. The next sections of this report briefly describe a typical process for identification of unspecified drug product impurities, and present an example of such an identification where the impurity turned out to be a leachable.
Identification Process for Unspecified Drug Product Impurities
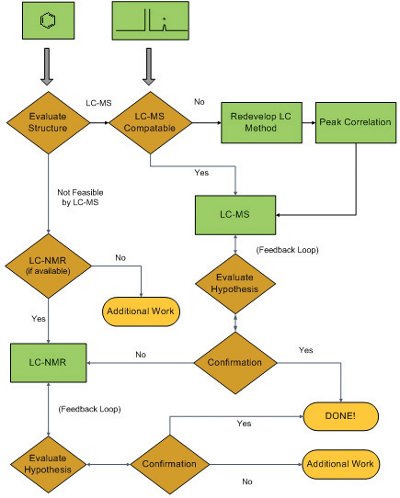
Figure 1 - Flowchart showing a typical drug substance/drug product unknown impurity structural analysis process
Pharmaceutical development laboratories that specialize in the structural analysis of drug substance/drug product impurities and degradation products often employ defined processes to facilitate this task [10, 11]. Such a process is presented as a flowchart in Figure 1. When an unspecified (i.e., previously unidentified) impurity is detected in an impurity profile (typically an LC/UV chromatogram), it is assumed that the new impurity is structurally related to the active pharmaceutical ingredient (API). For at least the past two decades, LC/MS has been the first-line analytical technique for structural analysis of pharmaceutical impurities, so the initial tasks are to evaluate the structure of the API in order to select the most appropriate ionization process (i.e., Electrospray (ESI) or Atmospheric Pressure Chemical Ionization (APCI); either one in positive or negative ionization mode), and to evaluate the LC platform/method for LC/MS compatibility (note that while it is possible to use LC methods with involatile mobile phase additives for LC/MS analysis, it is not desirable). In the unlikely event that LC/MS analysis is not feasible, many pharmaceutical development laboratories now have combined LC/ NMR (Nuclear Magnetic Resonance spectroscopy) with on-line solid phase extraction available. LC/MS and LC/NMR can generate structural hypotheses for subsequent confirmation. Such confirmation is accomplished by: 1) chemical synthesis of the proposed impurity and proof of its structure by NMR (supported by mass spectrometry and chromatography retention time match with the unknown in a real sample); or 2) demonstration through logical arguments that the LC/MS and NMR data available are sufficient to eliminate all other possible structures. In the event that LC/MS and LC/NMR data are not sufficient to generate structural hypotheses, additional work is required which typically involves isolation of relatively large quantities of the unknown impurity (i.e., several hundred micrograms to low milligrams) for more detailed NMR analyses and other analytical assessments (e.g., single crystal X-ray spectroscopy).
An Unspecified Drug Product Impurity – Is it a Leachable?
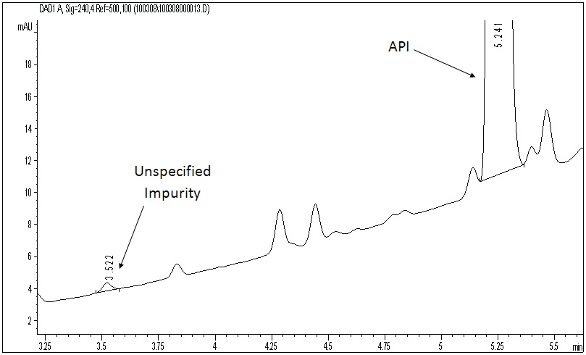
Figure 2 - LC/UV drug substance impurity profile (240 nm) showing the active pharmaceutical ingredient (API), a number of specified (i.e., known) impurities, and an unspecified (i.e., unknown) impurity (LC platform: C8 column, 4.6X150mm, 5μm particles; mobile phase A: 0.1% trifluoroacetic acid in water/B: 0.1% trifluoroacetic acid in acetonitrile; gradient: 30%B to 95%B in 10 min/ to 100%B in 3 min/hold for 3 min; flow 2 mL/min; column at 40ºC)
The following example case study is based on the authors’ professional experience and is designed to be representative of this type of pharmaceutical development problem. However, the sample under investigation in this case study is in no way related to products of the authors’ company (Boehringer Ingelheim Pharmaceuticals, Inc.) or any previous employer of any individual author.

Figure 3 - Diode array spectra of the API (top) and unspecified impurity (bottom)
Figure 2 shows an LC/UV impurity profile of a drug substance with an unknown impurity indicated, which was determined to be above the specified identification threshold. The diode array (DAD) UV spectra shown in Figure 3 indicate that the unknown impurity is structurally different from the API, suggesting that it is either a totally unrelated chemical entity or a very small molecular fragment with a significantly altered chromophore. Since the LC method was LC/MS compatible, LC/MS analyses were accomplished with both APCI and ESI processes with negative ion mode being the most sensitive for the unknown impurity in both ionization processes. Figure 4 shows a representative negative ion ESI mass spectrum, clearly indicating a nominal molecular weight of 190 for the unknown ([M-H]- at m/z 189). Further, the isotope pattern of the molecular ion indicates that the molecule contains two chlorine atoms. Since the API was known to contain no chlorine, it was hypothesized that the unknown impurity was a leachable of unidentified source which had migrated into the drug substance material at some point during its manufacturing process. Accurate mass measured LC/MS analysis (Figure 5) indicated a molecular formula for the unknown’s molecular ion of C7H3O2Cl2 (0.097 ppm mass accuracy), and LC/MS/MS analysis (Figure 6) with collision-induced dissociation showed a primary loss of CO2 from the molecular ion. The significant negative ion sensitivity in ESI along with the primary loss of CO2 suggested that the unknown was a carboxylic acid. This conclusion, considered along with the UV absorbance and chlorine content, further suggested that the unknown was an aromatic carboxylic acid with two aromatic substituted chlorine atoms. An isomeric dichlorobenzoic acid was consistent with all of the available mass spectrometric data on the unknown. LC/UV and LC/MS analyses on authentic reference compounds (e.g., Figure 7) confirmed that 2,4-dichlorobenzoic acid was the unspecified impurity. Since 2,4-dichlorobenzoic acid is not structurally related to the API, nor was it employed in the API synthetic process, it was confirmed to be a leachable and a search was initiated for its source.
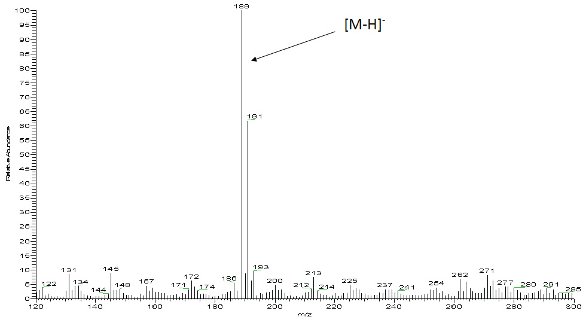
Figure 4 - Negative ion electrospray mass spectrum of the unspecified impurity
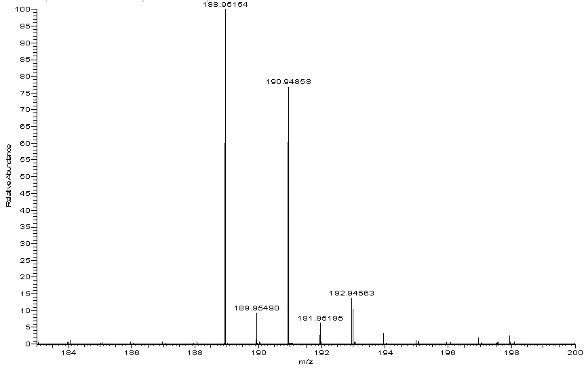
Figure 5 - High resolution accurate mass measured mass spectrum of the molecular ion region of the unspecified impurity (from an LC/MS analysis accomplished on an FTMS system)
Investigations subsequently determined that a silicone rubber tube attached to processing equipment used for the API synthesis was the source of the 2,4-dichlorobenzoic acid. It was determined that Bis(2,4- dichlorobenzoyl)peroxide was used for Vulcanization of the silicone rubber in that particular tubing: It should be obvious that Bis(2,4-dichlorobenzoyl)peroxide can easily degrade chemically to yield 2,4-dichlorobenzoic acid.

Figure 6 - Negative ion electrospray collision-induced dissociation mass spectrum of the unspecified impurity (from an LC/MS/MS analysis accomplished on an ion trap system)
Summary and Conclusions
In conclusion:

Figure 7 - LC/UV drug substance impurity profiles (240 nm) unspiked (top) and spiked (bottom) with 2,4-dichlorobenzoic acid
- Leachables can be an issue in low-risk dosage forms, such as solid orals and topicals.
- As with high-risk dosage forms, leachables in low-risk dosage forms can originate from a variety of sources and have a diversity of molecular structures.
- Leachables in low-risk dosage forms typically present as unspecified impurities in drug product impurity profiles.
- Unlike the situation with high-risk drug product types (e.g., OINDP, injectables, opthalmics) where Controlled Extraction Studies and leachables stability studies are accomplished during pharmaceutical development, “reverse-engineering” is the typical process used to determine the origin of leachables in low-risk dosage forms.
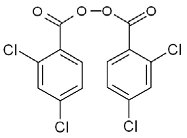
Structure 1
The reader interested in more detailed information regarding extractables and leachables, including origins, identification and analysis, and control, is referred to additional reports in the scientific literature [12- 24]. The reader should further note that as of this writing a PQRI working group is in the process of developing safety-based thresholds for leachables in parenteral and opthalmic drug products (PODP), as well as best demonstrated pharmaceutical development practices for these additional high-risk dosage forms (see: pqri.org). Also, the Extractables/Leachables Safety Information Exchange (ELSIE) is at work developing a database of safety information on known drug product leachables (see: elsiedata.org).
Acknowledgements
The authors acknowledge their employer Boehringer Ingelheim Pharmaceuticals, Inc. for their support, along with the Product Quality Research Institute, The International Pharmaceutical Aerosol Consortium for Regulation and Science (IPAC-RS), and the Parenteral Drug Association (PDA).
References
- Morton, M. (Ed.), Rubber Technology, Third Edition, Kluwer Academic Publishers, London, 1999.
- Container Closure Systems for Packaging Human Drugs and Biologics; Guidance for Industry; U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CEDER) and Center for Biologics Evaluation and Research (CBER); Rockville, MD, May 1999; 1-41.
- Metered Dose Inhaler (MDI) and Dry Powder Inhaler (DPI) Drug Products; Draft Guidance for Industry; U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CEDER); Rockville, MD, October 1998; 1-62.
- Nasal Spray and Inhalation Solution, Suspension, and Spray Drug Products – Chemistry, Manufacturing, and Controls Documentation; Guidance for Industry; U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CEDER); Rockville, MD, July 2002; 1-45.
- Norwood, D. L. (Chair), Product Quality Research Institute. September 2006. Safety Thresholds and Best Practices for Extractables and Leachables in Orally Inhaled and Nasal Drug Products. Submitted to the PQRI Drug Product Technical Committee, PQRI Steering Committee, and U.S. Food and Drug Administration by the PQRI Leachables and Extractables Working Group. http://pqri.org/pdfs/LE_Recommendations_to_FDA_09-29- 06.pdf (accessed 11/15/2006).
- Ball, D.; Blanchard, J.; Jacobson-Kram, D.; McClellan, R.; McGovern, T.; Norwood, D. L.; Vogel, M.; Wolff, R.; Nagao, L. Development of Safety Qualification Thresholds and Their use in Orally Inhaled and Nasal Drug Product Evaluation. Toxicological Sciences. 2007, 97(2), 226-236.
- Norwood, D. L.; Paskiet, D.; Ruberto, M.; Feinberg, T.; Schroeder, A.; Poochikian, G.; Wang, Q.; Deng, T. J.; DeGrazio, F.; Munos, M. K.; Nagao, L. M. Best Practices for Extractables and Leachables in Orally Inhaled and Nasal Drug Products: An Overview of the PQRI Recommendations. Pharmaceutical Research. 2008, 25(4), 727-739.
- Fang, X.; Cherico, N.; Barbacci, D.; Harmon, A.; Piserchio, M.; Perpall, H. Leachable Study on Solid Dosage Form. American Pharmaceutical Review. 2006, 9(7), 58, 60-63.
- Q3B(R) Impurities in New Drug Products; Guidance for Industry; U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CEDER); Rockville, MD, November 2003; 1-7.
- Norwood, D. L.; Qiu, F.; Mullis, J. O. Trace Level Impurity Analysis. Encyclopedia of Pharmaceutical Technology, 3rd Edition, Dekker Encyclopedias (a product line from Taylor and Francis Books), New York, pp. 3797-3813, 2006.
- Qiu, F.; Norwood, D. L. Identification of Pharmaceutical Impurities. Journal of Liquid Chromatography and Related Technologies. 2007, 30(5-7), 877-935.
- Norwood, D. L.; Prime, D.; Downey, B. P.; Creasey, J.; Sethi, S. K.; Haywood, P. Analysis of Polycyclic Aromatic Hydrocarbons in Metered Dose Inhaler Drug Formulations by Isotope Dilution Gas Chromatography/Mass Spectrometry. Journal of Pharmaceutical and Biomedical Analysis, 1995, 13(3), 293-304.
- Norwood, D. L.; Nagao, L.; Lyapustina, S.; Munos, M. Application of Modern Analytical Technologies to the Identification of Extractables and Leachables. American Pharmaceutical Review, 2005, 8(1), 78-87.
- Norwood, D. L.; Granger, A. T.; Paskiet, D. M. Extractables and Leachables in Drugs and Packaging. Encyclopedia of Pharmaceutical Technology, 3rd Edition, Dekker Encyclopedias (a product line from Taylor and Francis Books), New York, pp. 1693- 1711, 2006.
- Norwood, D. L. Understanding the Challenges of Extractables and Leachables for the Pharmaceutical Industry – Safety and Regulatory Environment for Pharmaceuticals. American Pharmaceutical Review, 2007, 10(2), 32-39.
- Ball, D. J.; Norwood, D. L.; Nagao, L. Utility and Application of Analytical and Safety Thresholds for the Evaluation of Extractables and Leachables in Drug Products. American Pharmaceutical Review, 2007, 10(5), 16-21.
- Mullis, J. O.; Granger, A.; Qin, C.; Norwood, D. L. The Analytical Evaluation Threshold (AET) Concept: Sensitivity and Analytical Uncertainty. Leachables and Extractables Conference Proceedings, Dublin, Ireland, March 5-6, 2008, Smithers Rapra Technology, Ltd., 4/1-4/20.
- Ball, D. J.; Norwood, D. L.; Nagao, L. PQRI: Safety Thresholds and Best Practices for Extractables and Leachables in Orally Inhaled and Nasal Drug Products. Leachables and Extractables Conference Proceedings, Dublin, Ireland, March 5-6, 2008, Smithers Rapra Technology, Ltd., 3/1-3/8.
- Norwood, D. L. Considerations for Outsourcing of Leachables and Extractables Testing. American Pharmaceutical Outsourcing, 2008, 9(2), 22-28.
- Norwood, D. L.; Mullis, J. O. “Special Case” Leachables: A Brief Review. American Pharmaceutical Review, 2009, 12(3), 78-86.
- Norwood, D. L.; Jenke, D.; Manolescu, C.; Pennino, S; Grinberg, N. HPLC and LC/MS Analysis of Pharmaceutical Container Closure System Leachables and Extractables. Journal of Liquid Chromatography and Related Technologies, 2009, 32, 1768-1827.
- Dohmeier, D. M.; Norwood, D. L.; Reckzuegal, G.; Stults, C. L. M.; Nagao, L. M. Polymeric Materials and Components for Use in Orally Inhaled and Nasal Drug Products. Medical Device Technology, 2009, 20(2), 32-38.
- Norwood, D. L.; Mullis, J. O.; Feinberg, T. N.; Davis, L. K. N-nitrosamines as “Special Case” Leachables in a Metered Dose Inhaler Drug Product. PDA Journal of Pharmaceutical Science and Technology, 2009, 63(4), 307-321.
- Norwood, D. L.; Ruberto, M.; DeGrazio, F. L.; Castner, J.; Wong, W. K.; Jenke, D. Extractables and Leachables from Rubber Materials Used in Pharmaceutical Systems. Rubber and Plastics News, June 29, 2009, 15-19.
Cristina Manolescu received her MS/BS in Chemical Engineering from Babeş Bolyai University, Cluj Napoca, Romania and works as an Analytical Scientist in the Analytical Development Function at Boehringer Ingelheim Pharmaceuticals, Inc. in Ridgefield, CT. She is a member of the Chemical Development Support group and her work focuses on in-process testing and characterization of drug substances and intermediates using multiple analytical techniques including HPLC, LC/MS, GC, and KF. Ms. Manolescu serves as a Board of Directors member for the Connecticut Separation Science Council.
Scott Pennino received his BS in Biochemistry from the University of New Mexico and works as an Analytical Scientist in the Analytical Development Function at Boehringer Ingelheim Pharmaceuticals, Inc. in Ridgefield, CT. He is a member of the Trace Analysis group and his work focuses on mass spectrometric characterization and structure elucidation of drug substances/ products, intermediates and degradation products using ultra high resolution LC/MS and GC/MS.
Daniel L. Norwood, Ph.D., is a Distinguished Research Fellow in the Analytical Development Function at Boehringer Ingelheim Pharmaceuticals, Inc. in Ridgefield, CT. He previously held positions at Magellan Laboratories, Glaxo Research Institute, Duke University Medical Center (Research Assistant Professor), and the University of North Carolina School of Public Health (Adjunct) where he received his Ph.D. He chairs the PQRI Working Group on Leachables and Extractables for orally inhaled and nasal drug products.