Abstract
This case study summarizes the expedient formulation approach taken to enable Phase I clinical trials of a new chemical entity (NCE) of a class known to have poor oral bioavailability in pre-clinical species. In two months, the project team was able to develop a resource-sparing solid formulation that overcame the Active Pharmaceutical Ingredient (API)'s slow dissolution kinetics in gastric pH and low solubility in intestinal pH. In vitro dissolution models in bio-relevant media were used to screen a series of formulations utilizing crystalline or amorphous API and select the most promising formulation candidates. Subsequently, in vivo animal models were used to select the one final solid formulation which showed approximately 2-fold exposure enhancement relative to a suspension-formulation benchmark. The selected dry blend of amorphous API and excipients could be filled into 001 capsules to supply 100 mg-strength dosage forms in the quantities required to conduct the high-dose first-in-human trials. With this formulation, this NCE became the first in its class to reach Phase I trials, demonstrate oral bioavailability and meet target efficacious exposure, thus achieving the pharmacokinetic proof-of-concept necessary to continue clinical development.
Introduction
Low solubility compounds of many classes of pre-clinical drugs face the uncertain fate of not having in-human oral bioavailability or requiring unpractical high doses for efficacy. In these cases, the project team has to balance speed to pharmacokinetic (PK) proof-of-concept (Phase I) with lineof- sight to the market formulation. This compound was being developed to provide a solid oral dosing alternative to the pertinent patient population. A target PK exposure was determined based on animal models of efficacy. Using in vitro - in vivo extrapolation (IVIVE), the daily dose needed to achieve target PK exposure was projected to be as high as 1300 mg. Due to high dose and low solubility, the probability of success of this molecule seemed low and, thus, pre-investment of resources (API, time and personnel) in market-like formulation development was not favored. This particular case study illustrates the approach taken to balance speed to Phase I with the goal of testing the oral bioavailability from a solid dosage form. A simple blend in capsule was developed to supply the clinical trials. To maximize the probability of success, in vitro models and animal in vivo models were used to screen multiple formulations, evaluate the physical form of the active ingredient and identify excipients, where all combined would most likely maximize PK exposure. When the most promising formulation candidates were identified, other criteria such as dosage size, ability to manufacture clinical supplies, and stability were factored in to select the final clinical formulation. Figure 1 depicts this step-wise screening and selection process.
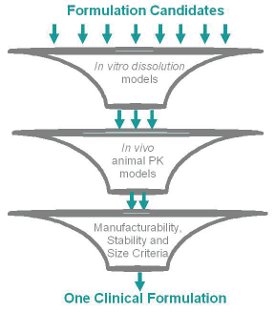
Figure 1 - Cartoon of the formulation screening and selection process1
Background
The compound is an ionizable molecule with three pKas. It was available as a stable crystalline salt with equilibrium2 solubility of 12 mg/ mL in Simulated Gastric Fluid (SGF) and 0.002 mg/mL in Fasted Simulated Intestinal Fluid (FaSSIF). At a 1000 mg dose, for example, the corresponding Dose Numbers [1] are then 0.3 (SGF) and 2000 (FaSSIF). The permeability of the compound, as determined by in situ rat intestinal perfusion studies, was estimated to be 6.42 x 10-5 cm/sec, which was higher than that of the high permeability reference compound metoprolol (3.69 x 10-5 cm/sec). Thus this compound was classified as highly permeable (BCS class II [1]). Due to high solubility in the SGF, it was expected that the dose will be solubilized in the stomach (dose number < 1). However, the apparently high solubility at gastric pH was counteracted by the slow dissolution of the neat crystalline compound in this medium; it takes more than 2 hours for the compound to dissolve (see Figure 2)3. Therefore the high dose was not expected to fully dissolve within the gastric residence time of ~ 30 minutes. The underlying cause of the slow dissolution was believed to be the kinetics of conversion to a crystalline hydrate of the same salt. The slow dissolution was seen as a liability to in vivo absorption, so, in an attempt to overcome it, the neat crystalline drug was rendered amorphous. Figure 2 shows a comparison of dissolution of neat crystalline and neat amorphous compound in SGF and FaSSIF. It was clear that the amorphous form of the compound dissolved faster than the crystalline form in the stomach pH thus making it more likely to have better bioperformance.
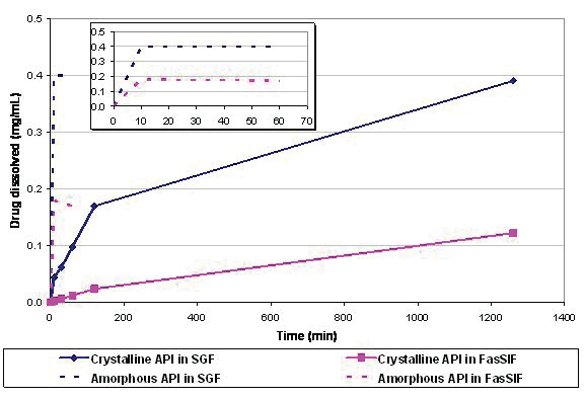
Figure 2 - In vitro dissolution of the neat crystalline API in one single medium (SGF or FaSSIF) at a total concentration of 0.4 mg/mL. Despite the favorable equilibrium solubility of the compound in SGF, complete dissolution of the crystalline material takes more than 120 minutes, which is longer than gastric residence time. In contrast, the neat amorphous API dissolves in 10 minutes (see inset). Similar behavior was observed at higher concentrations such as 4 mg/mL.
Once the dissolved drug transits into the intestine, the higher intestinal pH would likely cause precipitation due to low solubility under those conditions. This likely precipitation in the intestine was expected to be another liability to in vivo absorption. To mitigate this, antinucleating excipients were incorporated in the formulation for their potential to minimize or prevent precipitation of the dissolved API in intestinal conditions. In addition, surfactants were used to improve the wettability of the powders. Two-stage in vitro dissolution models were utilized to screen the formulations before testing them in animals. Twostage dissolution models attempt to mimic in a simple manner the time scale and sequence of pH and compositional environment the drug encounters in the gastro-intestinal (GI) tract. It is reported [2] that stomach medium presents an aqueous pH ranging from 1.5 to 3.5, containing no surfactant compounds, with liquid volume of 250 mL, and a residence time of approximately 30 minutes. The same source reports the intestine to present an aqueous pH medium ranging from 5 to 7, containing some surfactant bile salts in a total volume of approximately 500 mL and a resident time that may vary from 180 to 300 minutes. Two-stage method details can be found in the Experimental Procedures section.
Experimental Procedures
Formulation Methods
Limited amounts of API were allocated for development and only a total of eighty grams were used. Although multiple laboratory-scale formulation prototypes were tested in vitro, only three representative ones are discussed here. The formulation development was mainly aimed at enhancing the rate of drug dissolution and, hence, the pharmacokinetic exposure. The dissolution improvement was accomplished by rendering the API amorphous. Wetting agents (surfactants) and antinucleating excipients were added to increase the probability of success. Non-hygroscopic 001 capsule shells able to withstand storage with desiccant were used. One kind of formulation was made by encapsulation of a blend of the crystalline API with diluents, a polymer with potential antinucleating properties, a surfactant, a disintegrant, and a lubricant (Formulation 1). The second kind was made by encapsulation of a blend of the neat amorphous API with diluents, a polymer with potential antinucleating properties, a surfactant, a disintegrant, and a lubricant (Formulation 2). The neat amorphous API was prepared by spray drying it from an acetone solution of the crystalline powder. The resulting amorphous material had a glass transition temperature (Tg) greater than 160˚C. For both kinds, the final blend, containing approximately 25 % drug, was filled manually into capsules. For the third kind, a solid dispersion was made by spray drying an acetone solution of crystalline API and an appropriate polymer (at various drug contents, up to 50% API). Based on the high Tg and the need for high doses, a high drug content solid dispersion intermediate (50:50 API: polymer ratio) was blended with the antinucleating polymer and the surfactant, and then manually encapsulated to produce Formulation 3. The same surfactant and antinucleating polymer were used for the three formulations discussed here.
In Vitro Studies
All samples were tested with biorelevant media, such as Simulated Gastric Fluid (SGF, pH 1.8 HCl with 34.2mM NaCl) and Fasted Simulated Intestinal Fluid (FaSSIF, 28.7mM pH 6.5 Phosphate buffer with 6.53mM "crude" Sodium Taurocholate, 0.75mM Lecithin and 100mM KCl). These solutions were prepared according to references [3] and [4], where the FaSSIF was prepared at a higher concentration to solubilize all components and then diluted with water to 1X for single stage analyses or to 2X for the 2-stage analyses, where there is a 1:1 addition into SGF.
For the "neat" API dissolutions, approximately 5mg of the API was added to a glass scintillation vial containing 12mL of either SGF or 1X FaSSIF to create a concentration of ~0.4mg/mL. Samples were also prepared at 4mg/ mL in SGF and 2mg/mL in FaSSIF by adding 44 or 22mg to 12mL of media, respectively. These mixtures were stirred with a stir bar on a magnetic stir plate throughout the analyses. Samples were pulled manually by removing ~1mL with a plastic transfer pipette while stirring. The clarification [5] was performed by either centrifugation at 14,000RPM for 5 minutes (crystalline API) or at 80,000RPM for 8 minutes (amorphous API). The resulting supernatants were injected neat (crystalline API) or quantitatively diluted 1:1 with acetonitrile after centrifugation to prevent precipitation of supersaturated amorphous solutions during the chromatographic analysis.
For the 100mg API equivalent dosage form dissolutions, the analysis was performed at a larger scale than the above API experiments. A dissolution bath with paddles (USP II) at 100RPM was used. The volume was either 250mL of SGF or 500mL of FaSSIF, resulting in concentrations of 0.4mg/mL in SGF or 0.2mg/mL in FaSSIF. Procedurally, the dissolutions start in SGF for a specified amount of time, commonly 30 minutes. After the completion of this phase, 250mL of 37°C 2X FaSSIF (with a calculated yet small amount of concentrated sodium hydroxide to neutralize the SGF) was added to the vessel to transition the entirety of the vessel into the simulated intestinal conditions, resulting in a final volume of 500mL at pH 6.5. Samples were collected for a minimum of 90 minutes post FaSSIF addition. For all sampling timepoints, 3mL aliquots were pulled through a 10 μm flow filter. Amorphous formulations were further clarified by 80,000 RPM centrifugation for 8 – 10 minutes and diluted with acetonitrile in the same manner as the neat amorphous API experiment above.
Dog Studies and Pharmacokinetic Analysis
The formulations were tested in male Beagle dogs where the stomach pH was reduced to mimic human fasted stomach condition. Briefly, after an overnight fast, all dogs were treated intramuscularly with pentagastrin (0.006 mg/kg) approximately 30 minutes prior to dosing. After pentagastrin pretreatment, each dog was dosed the formulation (at 15 mg/kg) followed by 3.5 mL/kg water rinse via oral gavage. Water was restricted for 1 hour post dose. Food was returned at 4 hours after dosing. A 1-mL blood sample was drawn from a 21g catheter placed in the cephalic vein into EDTA tubes at pre-dose and 0.25, 0.5, 1, 2, 4, 6, 8, and 24 hours after dosing. The plasma was separated by centrifugation and kept frozen at –70ºC until analysis by liquid chromatography/tandem mass spectrometry. All animal studies were conducted under a protocol approved by the Merck IACUC.
Area under the curve (AUC0-24hr), observed maximum plasma concentration (Cmax), and time of Cmax (Tmax) were calculated using the linear trapezoidal, non-compartmental model in WinNonLin v5.2. Plasma concentration values below LOQ were set at zero for PK calculation purposes.
Results and Discussion
Figure 3 shows the two-stage dissolution profiles of the three representative capsule formulations. The two formulations containing amorphous API (2 and 3) approach or reach full dissolution at the 30 minute time point. In contrast, Formulation 1 containing crystalline API reaches less than 25 % dissolution at the same time point. After the pH, composition and volume of the media are changed to simulate the intestinal environment (FaSSIF), the API, as expected, tends to precipitate to meet its low equilibrium solubility (less than 1% on this graph). However, a significant portion of the drug released in SGF remains transiently2 solubilized in FaSSIF above equilibrium values over at least a two-hour period. These data provide strong evidence of the advantage of formulating the drug in the amorphous state rather than in the crystalline state. In addition, these data suggest that the antinucleating excipient may prevent the complete precipitation of the API.
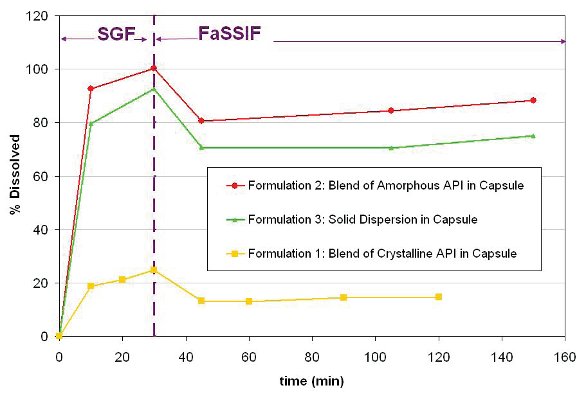
Figure 3 - Two-stage dissolution of capsule formulations. Here 100 % is 0.4 mg/mL (SGF) and 0.2 mg/mL (FaSSIF). Formulations 2 and 3 containing amorphous API reach or approach full dissolution at 30 minutes. In contrast, Formulation 1, containing crystalline API, releases less than 25 % over the same time period. In FaSSIF and over this time scale, the solubilized API remains above its equilibrium2 solubility of 0.002 mg/mL (or less than 1 % on this y axis scale).
Based on these in vitro results, Formulations 2 and 3 were chosen for a pharmacokinetic comparison in dogs against a benchmark suspension formulation of the crystalline API containing also an antinucleating polymer excipient with potential to maintain supersaturation in the intestinal environment.
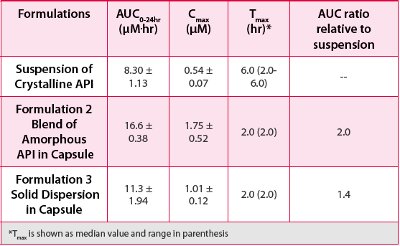
Figure 4 and Table 1 shows the average plasma concentration vs. time profiles and mean pharmacokinetic parameters, respectively, following oral administration at a dose of 15 mg/kg in pentagastrin pretreated dogs. The simple blend of amorphous API in capsule (Formulation 2) provided an approximately 2-fold exposure enhancement relative to the suspended crystalline API, with AUC0-24hr of 16.6 μm*hr vs 8.3. μm*hr, respectively. Also, these animal studies showed that the simple amorphous API blend had better bioperformance than the solid dispersion (Formulation 3), with AUC0- 24hr of 16.6 μm*hr vs 11.3 μm*hr, respectively. The PK data in dogs were consistent with the two-stage dissolution profiles in terms of rank-ordering of the formulations. The better bioperformance of the amorphous APIblend formulations compared with the solid dispersion and the crystalline benchmark is probably because it was able to dissolve more API in stomach, which then remains mostly solubilized in the intestinal fluid, hence achieving higher absorption.
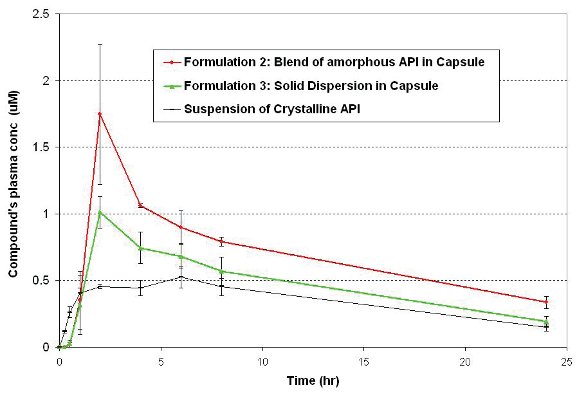
Figure 4 - Average plasma concentration profiles following oral administration at a dose of 15 mg/kg in pentagastrin pretreated dogs (n=3, mean ± SE).
Once this most promising formulation was identified, other considerations such as dosage form size, manufacturability and stability were factored into the final selection decision. With respect to size, this simple blend candidate could be filled into 00' capsules without densification to provide a 100-mg unit strength, which was adequate for incremental dosing up to the top Phase I dose. GMP encapsulation was feasible in a hand-operated filling system to support the Phase I trial quantity demands of the order of several thousands of dosage units. The stability predicted by accelerated studies at elevated temperatures indicated that the formulation would be chemically and physically stable if protected from moisture. Thus storage with desiccant was specified. To further mitigate the risk of unexpected instability, refrigerated storage (5˚C) was selected while long term stability studies were conducted concurrently with the clinical trial.
Table 1 - Average PK parameters following oral administration at a dose of 15 mg/kg in pentagastrin pretreated dogs (n=3, mean ± SE).
Clinical results following single oral dosing of GMP capsules of Formulation 2 in healthy subjects showed dose-proportional pharmacokinetic profile across the dose range with AUC0-∞ and Cmax linearly proportional up to the 1600-mg dose. Based on the oral bioavailability observed in these studies, it was predicted that the target exposure for efficacy would be achieved at doses lower than 500 mg, which was less than the 1300 mg predicted prior to the Phase I studies. This case study shows how a simple formulation approach complemented with in vitro and in vivo models of biopharmaceutical performance can help in expediting formulation development for first-inhuman trials.
Conclusions
In two months and with eighty grams of API, a solid dosage form of a low solubility compound was defined by utilizing in vitro and in vivo models to screen multiple formulations and select the one with the highest probability of providing enhanced pharmacokinetic exposure. The poor dissolution kinetics of the drug in SGF was overcome by spray drying the API to make it amorphous. The solid capsule formulation consisted of a simple dry blend of this amorphous API with excipients such as diluents, an antinucleating agent, a surfactant, a disintegrant, and a lubricant. The blend could be filled into 00' capsules to provide a 100 mg unit dose by utilizing manual GMP encapsulating equipment. This capsule formulation exhibited dose proportional pharmacokinetics in human and met target exposure at a dose lower than 500 mg, amply demonstrating the pharmacokinetic proof-of-concept required for this first-in-class compound to proceed further into development.
1 Although not used in this particular example, in silico models are often used to pre-screen formulations before the animal models.
2 Equilibrium solubility values were obtained by measurements at times greater than 24 hours. Due to dissolution kinetics and supersaturation, shorter measurements yield transient solubility numbers which may be higher or lower depending on the media pH relative to molecule's pKas and/or ability of the excipients to mitigate precipitation.
3 The crystalline API was milled and available as mostly primary particles of an average size of 24µm (SD = 13 µm, with 95% < 59 µm). The effect of further particle sizing was not explored.
References
- Gordon L. Amidon, Hans Lennernas, Vinod P. Shah, and John Crison, Pharmaceutical Research, Vol. 12, No. 3, 1995
- T. Kararli, Biopharm. & Drug Disp., vol 16, 351-380 (1995)
- Lardia-Arana, David; Kristensen, Henning G.; Mullertz, Anette. Journal of Pharmaceutical Sciences (2005), 95(2), 248-255
- Galia E, Nicolaides E, Horter D, Lobenberg R, Reppas C, Dressman JB. Pharm Res (1998), 15:698–705
- Harmon, Paul; Li, Li; Marsac, Patrick; McKelvey, Craig; Variankaval, Narayan; Xu, Wei. AAPS Newsmagazine (2009), 12(9), 14-20
Acknowledgements
We thank project team colleagues, Zhen Liu, Sachin Lohani, James Ormes and Matt Tudge for pre-formulation characterization and processchemistry contributions. We also thank Paul DeGregory for analytical support and Kim Manser and Becky Nissley for conducting the dog studies. We are grateful to Qing Chen, Wendy Comisar, Mark Mowery, David Storey, and Michele Trucksis for input and review.
Maria Teresa Cruañes obtained a Ph.D. in Physical/Analytical Chemistry from University of Illinois at Urbana-Champaign under Larry Faulkner and Harry Drickamer. She gained post-doctoral experience working with Eli Lilly and University of Kansas' Pharmaceutical Chemistry department under Val Stella. Maria has been with Merck at West Point, Pennsylvania, for 14 years. She has held various positions, first as a Scientist in Pharmaceutical Manufacturing, then as a Group Leader in Formulation R&D and, later, as a Group Leader in Analytical R&D, and currently, as Lean Six Sigma Lead in the Process Improvement Group in Pharmaceutical Sciences and Clinical Supply. She is Chair-elect of the newly formed "QbD and Product Performance" Focus Group of the American Association of Pharmaceutical Scientists.
Adam Socia is a Research Chemist in Pharmaceutical Sciences and Clinical Supply at Merck & Co., Inc. He holds a MS in Chemistry from Sacred Heart University and is working on his PhD in Analytical Chemistry at Drexel University.
Dr. Amitava Mitra is a Research Fellow in Pharmaceutical Sciences and Clinical Supply at Merck & Co., Inc. He holds a Ph.D. in Pharmaceutical Sciences from University of Maryland and post-doctoral fellowship from Fox Chase Cancer Center. He has published research and review articles in peer-reviewed journals as well as book chapters. He has numerous podium & poster presentations in national & international conferences.
Steven Hu obtained his Ph.D degree from Brown University majoring in Chemical Engineering. He worked in MIT as postdoctoral fellow for 2 years before joining J&J Pharmaceutical R&D. After that, he worked in Merck Research Labs for 2 years and half. He currently is the Director of New Product Development of GSK China R&D.