'If you don’t know where you are going, any road will get you there.' Lewis Carroll (1832-1898)
Introduction
In-licensing is perhaps the fastest growing component for augmentation of R&D pipelines in the pharmaceutical industry today. In-licensing opportunities can span the entire range of drug discovery, development, and commercialization and include proprietary research technologies designed to explore novel biological targets, promising but untested new chemical platforms, new compound entities (NCEs) at various stages of preclinical or clinical development, and marketed products. Due Diligence (DD) is a formal process for evaluating and assessing risk for prospective in-licensing opportunities by obtaining and examining information about the technical, financial, legal, and other material aspects of the other party and/or the specific asset(s) in which the buyer is interested; DD is a key step in ascertaining and understanding the risks associated with the asset and in determining what steps must be taken to mitigate those risks. In the DD process, the asset owner (seller) and purchaser (buyer) generally assemble separate out-licensing and in-licensing teams, respectively.
This manuscript focuses upon the DD evaluation of nonclinical safety risks of pharmaceutical assets. Usually a nonclinical safety specialist is on both the seller’s and buyer’s teams; sometimes these specialists are consultants under contract to one of the parties, especially if the team represents a small company. This manuscript is written from the perspective of the buyer because it is the buyer who knows the least about the asset and is generally working under strict time constraints to generate the knowledge to support a buying decision. However, this manuscript also has utility for the seller, especially if a seller has relatively little experience in selling a pharmaceutical asset, as it will help the seller understand what kinds of information the buyer will find particularly useful.
The risks associated with a pharmaceutical assets lie as much in delivery of enhanced disease management (or cure) and/or improved ‘synergies’ in patient care as in minimizing/avoiding development delays, clinical ‘holds’, strategic failures, and non-competitive product labeling. The nonclinical safety evaluation is focused exclusively upon the latter. The initial aim of the nonclinical safety evaluation is to identify: 1) Scientific findings that negatively impact development; 2) Technical issues that impact the validity of individual scientific studies, 3) Deviations from regulatory requirements and expectations that impact ‘fit for purpose’, and 4) Implications of findings for the product label that could potentially delay or derail product development and marketing authorization. As each of these risks is identified, the nonclinical safety evaluation broadens to include an assessment of the likelihood that the risk will be realized, and if the risk is realized an assessment of the magnitude of its impact, for example, project termination versus a 6-month delay to execute a mitigation plan.
Types of In-licensing Evaluations
The nonclinical safety evaluation of an in-licensing opportunity rarely begins with formal due diligence. Most often it begins with a non-confidential discussion between the seller and buyer, the goal of which is to establish the level of interest that both parties have in moving the deal forward. During this preliminary meeting the broad scope of the opportunity is communicated. In addition to high-level information about the pharmacological properties and intended indication, the discussion usually covers whether the opportunity involves just a single compound, or includes back-up compounds or other assets, and the current status of the discovery and/or development program. At this early point in the relationship information germane to the nonclinical safety specialist is generally quite cursory. The seller might, for example, make a statement that the nonclinical safety profile for the lead compound is acceptable. A statement of this kind invites questions that are reasonable even in the context of a non-confidential discussion. Which nonclinical studies have been completed? What types of findings are dose limiting? What are the primary organs of toxicity? The seller may not have the appropriate technical expert present at the preliminary discussion to answer these questions and may not be comfortable sharing details before a confidentiality agreement is signed, but at this early stage it is worthwhile to ask probing questions until these limits are realized. If there are nonclinical safety issues that will clearly preclude further development of the asset, it is better to identify them as early as possible because this can save everyone a lot of time working on a deal that is destined to fail.
If the non-confidential discussion between parties is successful, a subsequent and more detailed evaluation proceeds under a confidentiality agreement. For a compound that has entered, or is about to enter Phase I clinical trials the information made available by the seller typically includes an Investigator’s Brochure that contains high level summaries of the pivotal nonclinical studies that support the clinical trial application. While an Investigator’s Brochure is quite informative, many sellers employ a practice of making all nonclinical study reports available to the buyer in a secure electronic data room. A detailed assessment of the nonclinical safety profile is possible under these circumstances, and if an electronic data room is made available, the buyer should take full advantage of the opportunity for deep fact finding.
The final phase of in-licensing evaluation is a formal Due Diligence, which is the primary focus of this manuscript. The DD usually includes a one- to three-day visit by the buyer to the site at which the seller is located, but sometimes it is conducted virtually through electronic data rooms and teleconferences between the parties. The principal advantage of a site visit is that it permits face to face conversations between the analogous technical experts on the buyer’s and seller’s teams, and the value of these face-to-face conversations for ferreting out subtle interpretations of data sets should not be underestimated. A site visit also facilitates discussions between experts in different disciplines on the buyer’s team, the importance of which is noted in a subsequent section of this paper. Whether face-to-face or virtual, the primary purpose of DD from the buyer’s perspective is to verify that each of the claims made by the seller is supported by documented evidence. The goals are broader than simple verification of facts, and encompass establishing a clear understanding of the potential risks associated with the safety profile in terms of the probability that those risks will be realized and if so what their impact would be.
Different Nonclinical Evaluations for Different Development Stages
As stated above, in-licensing opportunities span the entire range of the pharmaceutical development life cycle – from target discovery to life cycle management. Different tools and non-clinical expertise should be considered, based upon the status of the opportunity. For early-phase opportunities there may be little internal documentation relevant to nonclinical safety, and the evaluation thus based upon what can be gleaned from published literature, government documents, and competitive intelligence. For later-phase opportunities, there may be a plethora of internal research reports, memos, submissions to regulatory authorities, communications with authorities regarding general regulatory and asset-specific issues; advisory panel transcripts, regulatory decisions, and litigation documents.
Discovery-Phase
A discovery-phase asset may be disclosure of a relationship between a novel molecular target and disease pathophysiology amenable to pharmaceutical discovery, technology with which to search for new chemical structural platforms, and/or chemistry (platforms and/or promising lead compounds). Depending upon how much effort the seller has invested in the opportunity, there may or may not be much internal documentation relevant to non-clinical safety for review. In these cases, the nonclinical safety investigation could consider public databases, published literature, data mining software, government documents, and competitive intelligence:
• Target and pathway evaluation:
- What is discernable about the molecular target or structurally similar targets from the literature (e.g. spontaneous models in which the target is implicated; genetic knock-out and knock-in models)?
- What intra- and inter-cellular pathways are linked to the target, and do any of those pathways have known safety implications?
- How is the target regulated in response to cell or tissue manipulations
- Is information available regarding competitor activities, and if so safety issues?
• Chemical platform and lead series structural risk – in addition to any documentation that may be provided (e.g. investigational safety pharmacology and toxicology studies), in silico analyses of the NCE’s structure can be conducted to evaluate potentials for mutagenicity and other properties of importance in toxicology for which the buyer has commercially available or customized proprietary predictive software.
• Metrics:
- How does asset measure up relative to buyer’s internal criteria for target selection and/or chemical lead molecule(s) at this stage?
Preclinical-Phase
 Generally, includes Discovery plus: i) a partial or complete first time in man (FTIM) package consistent with International Conference on Harmonization and regional guidance, ii) General toxicology & safety pharmacology risks sufficiently understood and with appropriate safety margins to support FTIM, and iii) pre-IND or pre-IMPD communications with regulatory authorities and meeting minutes (if available).
Generally, includes Discovery plus: i) a partial or complete first time in man (FTIM) package consistent with International Conference on Harmonization and regional guidance, ii) General toxicology & safety pharmacology risks sufficiently understood and with appropriate safety margins to support FTIM, and iii) pre-IND or pre-IMPD communications with regulatory authorities and meeting minutes (if available).
• Non-clinical Study Reports:
- Do the study reports accurately reflect the data contained therein?
- Are the experimental methods (in vivo, in vitro, analytical, and statistical) employed consistent with current standard practice, and if not are they sufficiently qualified (e.g. use of relevant pharmaceutical positive and negative controls) so that the results may be relied upon and/or defended if challenged by regulatory authority?
- Are there ‘outliers’ that could reflect potential risk not reflected in group-mean data? Are there any outlier responses (e.g. unscheduled death) in pharmacology studies (e.g. performance of the asset in an animal efficacy model) that could constitute a safety concern?
- Are the studies conducted by qualified individuals, and if conducted under contract for the Seller, by well-regarded contract research organizations?
• Regulatory nonclinical study reports: Regulatory reports are those upon which potential risks to human subjects participating in clinical trials will be based, and are require d to be conducted to Good Laboratory Practice (Code of Federal Regulations, Part 58) standards, or international equivalents (e.g., OECD, Directive 2004/9/EC and 2004/10/EC).
- Were the regulated studies conducted to the appropriate standards and is the supporting documentation present and ‘fit for purpose’ (see Table 1 for sources of region-specific information? Regulatory nonclinical studies not conducted to the appropriate standards and/or for which certificates are defective or missing may be required to be repeated before clinical trials or requests for marketing authorizations may proceed.
• Nonclinical safety studies cannot be evaluated in isolation, but must be evaluated within the contexts of: i) Preliminary maximum tolerable dose (MTD) and range-finding studies in the same species; ii) Subsequent (e.g. longer duration) studies in the same species; iii) Study designs and findings in the other species used in general toxicological studies to support the FTIM package; and iv) Study designs and findings in the non-general toxicological studies (e.g. safety pharmacology). In addition to adequacy of study designs, experimental findings, and compliance with standards and regulatory guidance:
- Are the doses and exposures to parent drug consistent across sets of studies in a given species? Across species?
- Is there experimental evidence that each study was dosed to achieve a maximum tolerated dose, and if not, are there any ‘exposure gaps’ which might contain new information relevant to patient safety if investigated?
- Is the design of the nonclinical studies consistent with, or a fair approximation of, how the drug will be dosed in the initial clinical program?
- Are there adequate safety margins for any serious nonclinical findings?
- Are there unusual data ‘outliers’ or other unexpected findings (e.g. unanticipated and/or unscheduled deaths) in any of the in vivo nonclinical studies (e.g. including nonclinical pharmacology/efficacy studies)?
• Metrics:
- From analysis of the non-clinical studies and summary documentation provided and the proposed clinical trial design, plus any additional information available to the reviewing nonclinical scientist (literature, government documents, competitive intelligence), is the toxicology coverage adequate to support Phase 1 clinical plan, and to address regulator concerns (if any)?
- Looking past the FTIM/Phase I clinical program, are there any ‘flags’ in the non-clinical data package that could signal pending difficulty to advance to Phase II/III or impede eventual product launch?
- How does the asset measure up against buyer’s scientific, regulatory, and commercial (if available) criteria for acceptability?
Generally, Discovery/Preclinical plus: i) Longer term non-clinical general toxicology; ii) Nonclinical safety investigative studies arising from results of nonclinical and/or clinical studies (if applicable); iii) Nonclinical studies to support inclusion of women of childbearing potential (WOCBP) in the clinical program and PIP plans; iv) Initiation of nonclinical carcinogenicity program, including submissions to FDA, FDA executive carcinogenicity assessment committee (CAC) recommendations, v) Pharmacokinetic and drug metabolism profiles in non-clinical species and humans (e.g. are previous safety margin estimates sufficient regarding potential human risks? Have disproportionate or unique human metabolites been identified?); vi) Ancillary non-clinical safety studies required before exposure of ‘large numbers’ of patients or as needed (e.g. photosafety, immunotoxicology, additional safety pharmacology studies), vii) Clinical safety data collected to date (e.g. adverse events [AEs], serious adverse events [SAEs]), and viii) Contents of Regulatory Q&A correspondence (which provide unique information as to regulatory agency concern and possible ‘experience’ with competitor submissions in the same or related areas).
• Metrics:
- From analysis of the non-clinical studies and summary documentation provided, plus any additional information available to the reviewing nonclinical scientist (literature, government documents, competitive intelligence) and clinical AEs/SAEs is the Toxicology coverage adequate to support Phase II/III clinical plan, and to address regulator concerns (if any)?
- Looking past the Phase III clinical program, are there any ‘flags’ in the non-clinical data package that could signal pending difficulty to advance the clinical program, or impede eventual Product Launch?
- How does the asset measure up against buyer’s scientific, regulatory, and commercial (if available) criteria for acceptability.
Phase III to Launch-Ready & Marketed Products
Generally, Discovery/Preclinical/Phase I-2 plus: i) Nonclinical chronic general toxicity, carcinogenicity, peripost natal and juvenile toxicology (if appropriate) ii) Nonclinical investigative and problem solving studies on matters arising; iii) Qualification of chemical specifications, including contaminants, degradation products, potential genotoxic impurities, and extractables and leachables (from packaging materials), iv) Clinical AEs/SAEs, and v) End of Phase II and pre-NDA meetings with regulatory authorities (briefing packages, correspondence, minutes, Q&As), annual IND updates and expedited reports to regulatory authorities.
For marketed products: NDA/MAA documents and regulatory correspondence, Advisory Committee correspondence, and annual product updates to regulatory authorities.
• Metrics:
- From analysis of the non-clinical studies and summary documentation provided and clinical AEs/SAEs, plus any additional information available to the reviewing nonclinical scientist (literature, government documents, competitive intelligence), is the toxicology program and knowledge generated ‘fit for purpose’ to support development for launch in intended global markets?
- Looking past the Phase 3 clinical program, are there any ‘flags’ in the non-clinical data package that could impede Product Launch or complicate toxicology support for life cycle management (e.g. new indications)?
- How does the asset measure up against buyer’s scientific and regulatory criteria for acceptability?
Interdisciplinary Communication
For most in-licensing opportunities the buyer will assign a cross-functional team of technical experts to conduct the in-licensing evaluation. In order to conduct a comprehensive evaluation the nonclinical safety expert on this team needs key information from experts in other disciplines. These interdisciplinary lines of communication are not limited to those within the buyer’s team of which the nonclinical safety expert is a member, but should also include contacts with the corresponding experts on the seller’s team. Some Examples:
- An expert in pharmacokinetics can provide insights on exposure to the parent molecule and metabolites in nonclinical safety studies as well information on as protein binding, reactive metabolites, and possible species differences in metabolism. Understanding ho w the metabolite profile in humans is similar or different to that for the species used in safety studies is critical to assessing whether exposure to the metabolites in safety studies adequately includes assessment of the potential adverse effects associated with metabolites.
- The individual responsible for investigating chemistry and manufacturing controls (CMC) can provide important details about the specifications for impurities, especially impurities that are potentially genotoxic. These details are necessary to determine whether the impurities have been properly qualified in nonclinical safety studies.
- The clinician on the team should be consulted on details about AEs/SAEs and future clinical plans. The important aspects include age, sex and reproductive status of volunteers or patients, duration of clinical trials, route of dosing, and dosing regimen. These clinical features are important for assessing whether the nonclinical safety program will support the clinical plans or will have to be augmented with additional safety studies before the clinical plans can be implemented. The clinical expert also examines safety data collected during clinical trials with volunteers and patients in detail. A discussion between the clinical and nonclinical safety experts about adverse events that are dose limiting and similarities and differences between the clinical and nonclinical safety profile can provide insight about the extent to which the nonclinical safety findings translate to humans.
- During the evaluation the regulatory affairs specialist will be looking at all records of communications with regulatory authorities, such as minutes of meetings and written correspondence, and these records often contain items of interest for the nonclinical safety expert. The correspondence can provide important insights about any concerns the regulators might have about the nonclinical safety profile of the drugs under investigation.
Although interdisciplinary conversations between the nonclinical safety expert and experts in pharmacokinetics, CMC, clinical, and regulatory affairs are consistently useful from one in-licensing evaluation to another, each asset under evaluation is unique and the nonclinical safety expert should establish clear communication with any other disciplinary expert as circumstances dictate. For example, the nonclinical toxicology data might have clear implications for restrictions concerning use in certain patient populations, such as children or women of child bearing potential, or it might reveal gaps in the nonclinical safety program that require significant time to rectify. These potential restrictions or delays to launch could significantly impact the commercial forecast for the asset and thus need to be promptly communicated to the expert responsible for valuing the asset.
Identifying Key Safety Issues and Risks
A thorough nonclinical safety evaluation will confirm whether all the studies required or expected for the given stage of discovery or development are present. More important however, is developing an appreciation of the implications of those findings for humans enrolled in clinical trials and patients who will use the drug in therapy. The evaluation should list the treatment-related findings for each nonclinical secondary pharmacology, safety pharmacology, and toxicology study in the program and note the exposure (plasma Cmax and or AUC) of the parent and any metabolites associated with those effects, as well as the No-Observed- Adverse-Effect Level (NOAEL). We provide a template (Table 3) for consistent collection of individual nonclinical study information. Relationships between different types of findings, such as linkage between plasma clinical pathology and anatomic pathology findings or relationships between in vitro and in vivo results, such as inhibition of the hERG ion channel in vitro and QTc prolongation in vivo, and relevant safety margins should be highlighted.

With the complete list of safety findings in hand, the nonclinical evaluator must make a professional judgment about the likelihood that the findings will occur in humans. It is beyond the scope of this paper to describe the details of making this judgment, but a common approach relies on computing safety margins. Different companies compute safety margins in different ways, but they generally involve the ratio of plasma exposure (Cmax and or AUC) in animals at the NOAEL dose for each finding and the estimated or known plasma exposure in humans at the anticipated therapeutic dose. Corrections for protein binding are often used to account for species differences in the amount of unbound drug that is available to interact at the molecular targets of toxicity. Margins based on Cmax are generally used for adverse effects that are expected to vary with dynamic (e.g. moment to moment) variation in exposure, such as pharmacologically-driven changes in blood pressure, whereas margins based on AUC are used for anatomic pathology findings that are more likely to be driven by accumulated wear and tear, in which case a time-integrated metric such as AUC is regarded as more appropriate.
All drugs are associated with some risk, and small safety margins that approach unity generally mean higher risk. One exception to this generality involves findings that are regarded as specific to a single non-human species, in which case the evidence supporting species specificity must be clear and compelling (usually based upon a specific mechanistic argument). There is no simple rule for determining how large a margin needs to be for the risk level associated with a particular type of safety finding to be acceptable. Nonclinical safety findings that occur in multiple species, show no evidence of recovery, are life-threatening or pose significant medical complications, and cannot be monitored in volunteers or patients clearly demand a greater safety margin than findings that have none of those characteristics. How much risk a patient might tolerate is also dependent on the risk/benefit analysis; patients and physicians may accept a greater risk of adverse side effects (lower safety margins), if the treatment offers substantial benefits in treating a serious disease for which current therapies are not adequate.

When all the risks identified by review of the nonclinical safety data are understood, the next step is to assess the impact that such findings might have if they actually occur in humans. A mitigation strategy for each risk should be developed. Mitigation plans may include actions such as cautious dose escalation steps during initial clinical trials, monitoring of safety biomarkers predictive of adverse effects, and the availability of medical intervention to counteract the adverse effect if it should appear. Certain types of nonclinical findings, were they to appear as frequent adverse effects in humans (e.g. seizure), are frankly unacceptable from a patient safety perspective, and the impact of realizing those risks is termination of the project. All high probability risks in this category must be communicated to decision makers on the buyer’s team.
Understanding What is Absent in the Nonclinical Safety Program
Although evaluating what has been done in nonclinical safety studies and constructing a risk assessment for the findings are key objectives, developing an appreciation of what is not present, but should be, is equally important. Unless the asset already has marketing approval, it is critical to note all the nonclinical studies that remain to be conducted before the marketing application can be filed. The list should include how long the studies will take to complete and a rough estimate of cost. The nonclinical safety specialist also needs to determine if additional work must be done to make the asset ‘fit for purpose’ relative to the immediate expectations and plans. For example, if the asset is claimed to be Phase II –ready and the buyer’s clinical team wants to start a 8-week Phase II clinical trial immediately after the asset is acquired, but the existing toxicology studies only provide coverage for clinical trials that are 4 weeks in duration, this gap must be highlighted so that time needed to complete longer toxicology studies can be factored into buyer’s overall evaluation of the asset.
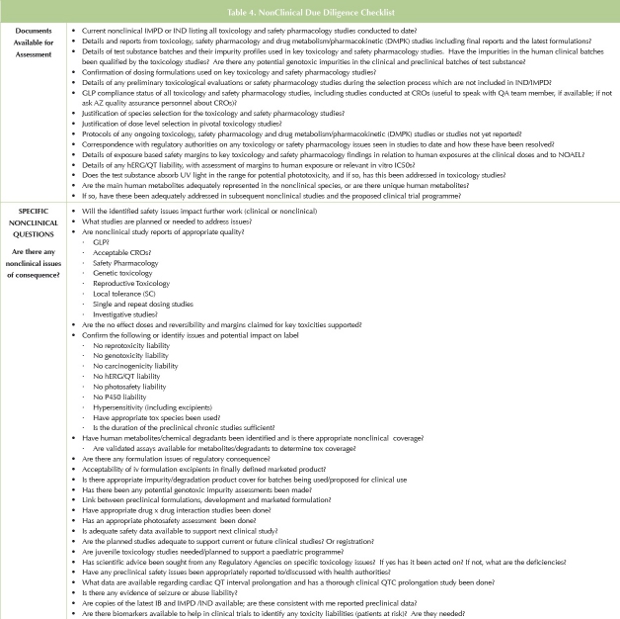
For projects that have advanced into late Phase 2 or beyond, several nonclinical safety testing issues are often underestimated or missed entirely by sellers, particularly if the seller is a small company with relatively little experience. One frequently overlooked issue pertains to impurities. The seller should have a clearly specified procedure for determining whether impurities are potentially genotoxic by in silico analysis and whether they need to be tested by in vitro genotoxicity procedures. Finding out late in development that an impurity has to be controlled to a less that a few ppm instead of 0.1% because it is potentially genotoxic can pose very significant manufacturing challenges that could take years to resolve. Qualification studies for human-specific metabolites can be complex and resource and time intensive. Evaluation of the potential for metabolites to form reactive intermediates should be available and the safety implications of any reactive metabolites should be carefully considered. Sometimes a seller will not appreciate that a significant change to the drug product late in development, such as free base to a salt form, will probably trigger the need for bridging toxicology studies to ensure that the toxicity profile has not concomitantly changed. The adequacy of bridging studies should be carefully interrogated.
Another common risk is that specific nonclinical studies required to advance clinical development or marketing authorization are missing or not adequately performed to be ‘fit for purposes’ leading to delays to conduct/repeat the study . The assessor must have a thorough and current working knowledge of regulations and regulatory guidance for all nonclinical studies necessary to support clinical trials and marketing authorizations (see Table 2 for a comprehensive listing), and of the types of documents containing non-clinical information that sponsors submit to regulatory agencies.
It is also important to determine that impurities in the drug substance to be evaluated in humans have been qualified in the general toxicity studies. As indicated earlier this most often occurs when changes to the synthetic process are made late in the development program. If this happens it is essential for the assessor to work with his/her CMC colleague to ascertain if new impurities are now present and require qualification.
Photosafety testing requirements are inconsistent in different regulatory regions, although ICH guidance is in development. The nonclinical safety evaluation should determine whether photosafety has been assessed at all, and if so, how the assessment will be regarded by regulators in the global regions where the buyer plans on launching the drug.
Have a Plan/Know What to Look For
Lewis Carrol’s aphorism is particularly apt for nonclinical DD; the nonclinical safety assessment requires that the assessor: 1) have a ‘road map’; 2) Take a prospective approach to what to look for in terms of non-clinical risks to the asset; and 3) Have strategies for finding them. Each DD is different, depending upon the maturity of the opportunity, the breadth and depth of investigations and information available, and the organizations and personnel involved. In preparing a plan for DD, we have developed a ‘check list’ of questions to assist in the non-clinical assessor as to where to focus, particularly with regard to deficiencies (Table 4).
Finally, sponsors should seek assurance from sellers and no individuals on FDA's List of Debarred Persons have been involved in any aspect of product development, including nonclinical studies or summary documentation. For products to be launched in the US, sponsors are required to certify that no person currently on the FDA’s List of Debarred Persons participated in any way with nonclinical aspects of the project’s development/NDA activities. (see http://www.fda.gov/ICECI/EnforcementActions/FDADebarmentList/default.htm?utm_campaign=Google2&utm_source=fdaSearch&utm_ m e d i u m = w e b s i t e & u t m _ t e r m = L i s t % 2 0 o f % 2 0 D e b a r r e d & u t m _ content=1)
The Risk is High (if such a person was shown to be involved – all work potentially associated with that individual would be challenged or disallowed, jeopardizing a launch scheduled; however, Probability is Low, as the individuals on FDA’s List are primarily physicians who were involved in fraudulent clinical trial activity.
Conclusions
The non-clinical assessment in the DD process is both detailed and complicated, and can be of great significance to identify potential risks to the asset and mitigation strategies. The assessment should be undertaken only by individuals experienced with the current and future drug development phases of the asset. Finally, because the DD process almost always takes place within a fixed time period, it is critical that the non-clinical assessor be free of competing responsibilities to devote full attention to the evaluation, and be guided by a prospective nonclinical assessment plan.
Dr. Lewis Kinter is currently Senior Director: Regulatory Toxicology, and Head: Toxicological Operations, Safety Assessment (US) at AstraZeneca Pharmaceuticals in Wilmington, PA., and manages preclinical safety programs conducted in support of AstraZeneca’s pharmaceutical clinical development activities in the US. Dr. Kinter has been engaged in pharmaceutical research and development and comparative physiology/medicine for 30 years and is an internationally recognized expert in cardiovascular-renal physiology, pharmacology, and toxicology.
Dr. Greg Christoph is Senior Project Director at AstraZeneca in Wilmington, DE. He leads a group of safety assessment professionals who design and deliver nonclinical safety testing programs for candidate drugs across multiple disease areas. Dr. Christoph has directed and guided the nonclinical contributions for successful INDs and NDAs for global regulatory purposes. In addition to coordinating the activities of scientists who provide nonclinical safety inputs to all in-licensing projects at AstraZeneca, he has conducted dozens of in-licensing evaluations for small molecules at all stages of drug discovery and development. Prior to joining AstraZeneca, Dr. Christoph held various research and leadership positions at DuPont, a major chemical company.
Dr. JoAnne Saye received her Ph.D. in Pharmacology from the University of Virginia. She has worked in the pharmaceutical industry for over 20 years, working at AstraZeneca Pharmaceuticals LP and DuPont Merck/DuPont Pharmaceuticals. Dr. Saye has provided toxicological support to development and discovery projects for AstraZeneca as well as participating on project teams. She has also participated in many due diligence opportunities, evaluating the non-clinical safety of compounds across most therapeutic areas. Her major responsibility at DuPont Merck/DuPont Pharmaceuticals was general pharmacology/safety pharmacology studies. Dr. Saye is a former chair of the General Pharmacology/Safety Phamacology Group (now the Safety Pharmacology Society). She is also active in the New York Academy of Sciences, having served as chair of the Biochemical Pharmacology Discussion Group from 1997-2001.