Abstract
Ligand binding assays (LBAs) are the primary methods used to quantify biotherapeutics, biomarkers and anti-drug antibodies to support biotherapeutics development. LBAs have been proven more difficult to outsource owing to their unique challenges such as high assay variability, nonlinear calibration, narrow dynamic range, more matrix interference, etc. Changes in reagents, instrument, bioanalyst, lab environment (temperature, humidity, etc) during assay transfer can have profound effect on assay performance in sensitivity, specificity, selectivity, and reproducibility, etc. Therefore, special attention to assay details is crucial for successful LBA transfer from lab to lab to ensure reproducible results to support regulatory submissions.
Biotherapeutics including peptides, recombinant proteins, monoclonal antibodies, oligonucleotides, etc. have gained tremendous growth in recent years and this trend will continue for the foreseeable future. Over the last several years, global market for biologics or biotechnology drugs increased dramatically and reached $120 billion in 2008 [1] and experienced double-digit growth each year, far more than one would have predicted a few years ago. Many major pharmaceutical companies that have historically focused on small molecule drugs have now allocated considerable budgets and technical resources to develop biologic products internally or through external collaborations. The spending on biologic drugs is growing nearly twice as quickly as spending on traditionally developed small molecule drugs. It is predicted that by 2014, the biggest-selling medicines will be biologics, according to analysis, and half of the top 100 drugs in 2014 will be biotech medicines with a total sale predicted to be ~$169 billion [2].
Reliable bioanalytical methods are the foundation for generating pharmacokinetic (PK), pharmacodynamic (PD) and immunogenicity data to support biotherapeutics development and registration. Ligand binding assay (LBA) is the primary method used for quantification of biotherapeutics, biomarkers and anti-drug antibodies (ADA) in biologic matrices to support PK and PK/PD evaluations during biotherapeutics development and market approval. The necessity for these assays is rapidly growing as the increase of biotherapeutics in development and the growing need for biomarker assays to assess therapeutic efficacy. While chromatographic assays are relatively less challenging and have already been routinely outsourced to contract research organizations (CROs), LBAs, however, have proven more difficult to develop and to outsource due to their unique challenges that require specific considerations for outsourcing.
LBA, as opposite to chromatographic assays that are primarily used for low molecular weight drugs bioanalysis, do not directly measure the analyte of interest but indirectly measure the binding interactions between the analyte and its specific reagents in the presence of other proteins such as serum factors, soluble receptors, endogenous molecules and/or structural analogs in biological matrices. LBA has evolved over the last several decades from radioimmunoassay (RIA) to a wide range of highly sensitive non-radioactive binding assays that rely on high-specific-activity nonisotopic labels.  The most commonly used LBAs include enzyme-linked immunosorbent assay (ELISA or EIA) and its variations such as fluorescence immunoassay (FIA), chemiluminescence immunoassay (CLIA), time-resolved fluorescence assay (TRF) and electrochemiluminescence assays (ECL). An example of a sandwich ELISA is graphically presented in Figure 1 where the analyte (a mAb as an example) is captured by a capture antibody and detected by an enzyme-labeled detection antibody. Unbound analyte and other matrix components are removed from the assay by repeated washing and the bound analyte is then detected via an enzyme-catalyzed color reaction. Other binding assays such as surface plasmon resonance (SPR) immunoassay, hybridization assay, immuno-PCR assay, bioassay, etc. have further expanded the applications of LBA [3].
The most commonly used LBAs include enzyme-linked immunosorbent assay (ELISA or EIA) and its variations such as fluorescence immunoassay (FIA), chemiluminescence immunoassay (CLIA), time-resolved fluorescence assay (TRF) and electrochemiluminescence assays (ECL). An example of a sandwich ELISA is graphically presented in Figure 1 where the analyte (a mAb as an example) is captured by a capture antibody and detected by an enzyme-labeled detection antibody. Unbound analyte and other matrix components are removed from the assay by repeated washing and the bound analyte is then detected via an enzyme-catalyzed color reaction. Other binding assays such as surface plasmon resonance (SPR) immunoassay, hybridization assay, immuno-PCR assay, bioassay, etc. have further expanded the applications of LBA [3].
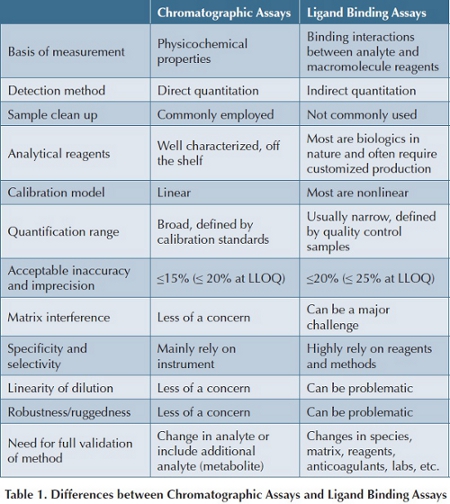
Although efforts have been made to explore different methodologies for quantitation of biotherapeutics with the advance of new technologies, LBA remains to be the preferred choice for biotherapeutic quantitation owing to its sufficient sensitivity, minimal requirements for sample cleanup, high sample throughput and relatively low per sample cost. Regardless of different formats of LBAs, they share some common characteristics that are very different from chromatographic assays. The major differences between LBAs and chromatographic assays along with their unique challenges to outsourcing are summarized in Table 1, and some of them will be discussed briefly along with some examples [4,5].
Assay Reagents
One of the key differences between LBAs and chromatographic assays is assay reagents. For chromatographic assays, the most commonly used reagents are organic solvent and buffers. They are not only readily available off the shelf but are also well characterized. Batch-to-batch or lot-to-lot variations in quality are normally not expected. However, LBAs not only rely on specific reagents (i.e., target antigen, capture or detection antibody, etc.) to the analyte of interest but often some of these reagents are proprietary assets, not readily available off the shelf and require customized design and production. The design, production, characterization and selection of specific assay reagents are often time consuming and labor intensive. Most importantly, since these reagents are often derived from biological sources (e.g., animals, cell lines, and micro-organisms, etc.), batch-to-batch and lot-to-lot differences are expected. Production of analyte-specific reagents is often at certain risk of not achieving the desired characteristics following a long production and selection processes resulting in repetition of the entire reagent production and selection process. Thus, ensuring enough supply of key reagents from the sponsor is crucial for a CRO to re-establish the analytical method and successfully complete method validation and sample analysis [6]. Even though some of the commonly used reagents for LBA (e.g., capture or detection antibody against therapeutic human IgG monoclonal antibody) can be purchased directly from different vendors, vendor-to-vendor and lot-to-lot differences have been found to have dramatic effect on assay performance due to potential differences in reagent specificity, binding affinity, specific activity of label, etc. Differences in assay performance due to differences in reagents supply are commonly observed by analysts during method development and assay transfer from lab-to-lab. Information sharing and ongoing communications between sponsor and CRO are important to avoid the CRO to perform unnecessary method development and/or reagent selection work that has been investigated by the sponsor.
Matrix Interference
Another major difference between chromatographic assays and LBAs is matrix interference. Most biotherapeutics are peptides, proteins or oligonucleotides in nature. They are sensitive to temperature, pH, ionic strength, and organic solvents. Typical extraction methods used for small molecule drugs via protein precipitation, liquid-liquid extraction or solid phase extraction in organic solvents are not suitable for biotherapeutics. 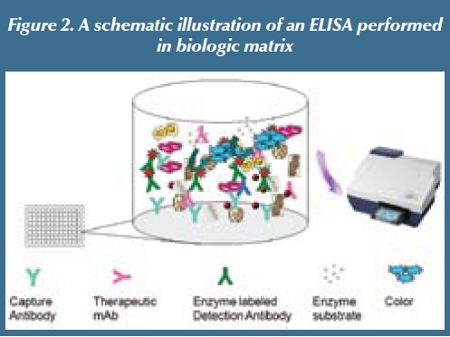 Therefore, biotherapeutic analytes generally do not undergo separation from other matrix components prior to analysis using LBA. As such, LBA development often encounters one of the major challenges resulting from cross reactivity or nonspecific interactions between analyte, and other molecules existing in complex biologic matrices [7], leading to either enhancement or suppression of binding signals. Thus, LBA relies on highly specific reagents to be able to distinguish the analyte from its degradation product(s), concomitant medication(s), and other endogenous counterpart molecules (e.g., therapeutic mAb versus endogenous IgG) that coexist in biological matrices. Even with highly specific reagents, matrix interference sometimes may still not be avoidable due to the presence of endogenous counterpart(s) or other unrelated matrix components, such as from homolysis, serum proteins, lipemia, etc. A schematic illustration of biologic matrix interference of LBA is presented in Figure 2.
Therefore, biotherapeutic analytes generally do not undergo separation from other matrix components prior to analysis using LBA. As such, LBA development often encounters one of the major challenges resulting from cross reactivity or nonspecific interactions between analyte, and other molecules existing in complex biologic matrices [7], leading to either enhancement or suppression of binding signals. Thus, LBA relies on highly specific reagents to be able to distinguish the analyte from its degradation product(s), concomitant medication(s), and other endogenous counterpart molecules (e.g., therapeutic mAb versus endogenous IgG) that coexist in biological matrices. Even with highly specific reagents, matrix interference sometimes may still not be avoidable due to the presence of endogenous counterpart(s) or other unrelated matrix components, such as from homolysis, serum proteins, lipemia, etc. A schematic illustration of biologic matrix interference of LBA is presented in Figure 2.
The degree of matrix interference varies depending on the analyte, reagents, biologic matrix and the analytical method. Sources, quality and disease stage of biologic matrices may have profound effects on assay performance. For example, during one method transfer, the recovery of analyte in control plasma samples was acceptable before transfer based on the specific a priori acceptance criteria (80-120%) of the nominal values in more than 80% of independent sources of the same matrix). After transfer, however, the recovery of the analyte evaluated at the CRO in 12 individual lots of plasma samples ranged from 21.3 to 192% and only ~42% of the lots (5/12) had a recovery within ± 20% of its nominal value. Following investigation, it was noticed that various degrees of homolysis exist in some of these individual plasma samples and they have likely contributed to the poor recovery of the analyte in some of the plasma samples. Additionally, selection of blank biologic matrix may have direct impact on assay performance. Difference in the blank matrix responses (backgrounds) between calibration standards and quality control samples could lead to repeated assay failure. Furthermore, differences in processing of biologic matrix have also been found to have significant effects on assay performance. For example, some labs centrifuge biologic samples prior to analysis while other labs may not do so. The difference in removing more or less matrix interference during sample preparation could be significant enough in altering assay performance based on our past experience, especially when study samples are prepared from one lab and calibration standards are prepared from different labs using different processes. Last, but not the least, is the impact of disease state on assay performance. A fully validated assay using control matrix from healthy subjects may not function well during sample analysis due to increased interference from elevated therapeutic ligand, analyte degradation product(s) and/or ADA in incurred samples. Therefore, understanding the intended use of analytical methods from the sponsor during early method development is crucial for a CRO to provide successful bioanalytical support to the program.
Nonlinear Calibration Curve
 Traditional LBAs have a narrow (1-2 orders of magnitude) and nonlinear calibration range that is most often fitted to a 4- or 5-parameter logistic model [8]. Quantification range of LBA is defined by validation samples or quality control (QC) samples based on their performance during method validation, which is different from chromatographic assays that employ calibration standards to define quantification range. Because of the heterogeneous nature of both biotherapeutics and their binding reagents, potential interference from matrix components and the multi-step nonlinear binding interactions between analyte and reagents, LBAs tend to have poor batch-to-batch or plate-to-plate reproducibility compared to chromatographic assays [9]. Replicate samples are typically measured to improve assay accuracy. Different labs have different preferences on how to integrate data from replicate samples when extrapolating concentration data from calibration curves: some labs may choose to average replicate response values first and then extrapolate the mean response value from calibration curve while other labs may prefer to extrapolate analyte concentration from individual response value and then average the concentration data. Although both approaches are acceptable based on current industry practice, different approaches could generate different concentration values.
Traditional LBAs have a narrow (1-2 orders of magnitude) and nonlinear calibration range that is most often fitted to a 4- or 5-parameter logistic model [8]. Quantification range of LBA is defined by validation samples or quality control (QC) samples based on their performance during method validation, which is different from chromatographic assays that employ calibration standards to define quantification range. Because of the heterogeneous nature of both biotherapeutics and their binding reagents, potential interference from matrix components and the multi-step nonlinear binding interactions between analyte and reagents, LBAs tend to have poor batch-to-batch or plate-to-plate reproducibility compared to chromatographic assays [9]. Replicate samples are typically measured to improve assay accuracy. Different labs have different preferences on how to integrate data from replicate samples when extrapolating concentration data from calibration curves: some labs may choose to average replicate response values first and then extrapolate the mean response value from calibration curve while other labs may prefer to extrapolate analyte concentration from individual response value and then average the concentration data. Although both approaches are acceptable based on current industry practice, different approaches could generate different concentration values.
As most LBAs have a nonlinear analyte concentration-response relationship, hook effects may exist at the upper end of the curve, resulting in a significant underestimation of drug concentrations when test samples have relatively high exposures. Since LBAs are often sensitive enough to measure picomolar or subnanomolar drug concentrations and have a narrow calibration range, samples with higher drug concentrations are often subject to hundreds- or thousands-fold dilutions prior to analysis using LBA. Dilution linearity is particularly important for LBA in order to demonstrate the validity of the method to generate reliable data. In an example from a past experience, a validated method was transferred to a CRO and successfully re-established and validated in the lab prior to sample analysis. However, analytical data indicated that some of the drug levels were significantly lower (~50-100 fold) than one would predict based on the known PK of the molecule and its dosing regimen. Close examination of dilution factors and data entry did not find any human errors. Further evaluation of each analytical run indicated that all analytical runs are acceptable based on the performance of both calibration standards and QCs. However, when close attention is paid to the shape of calibration curves employed during sample analyses, it was discovered that the responses between high QC (HQC) and upper limit of quantitation (ULOQ) of the curve appear to plateau and all suspected samples had a response value within this range of the curve. Repeat analysis of these samples with adequate dilution (50-200 fold) achieved quantifiable levels that are consistent with their PK profiles. This example highlighted the challenges for CROs to estimate sample dilution factors prior to analysis or recognize similar issues when project information (e.g., dosing regimen, route of administration, pharmacokinetic in relevant species, etc.) is not available.
Assay Robustness and Ruggedness
LBA consists of multiple steps and are affected by muitiple factors. Assay optimization requires multifactoriral consideration since changing of one assay condition may require re-optimization of other assay conditions and impact the overall performance of the assay. Evaluation of each assay condition such as reagent concentration, incubation time, temperature, etc. under various conditions are time consuming and labor intensive and may not be feasible during method development. For a given method, it may be more sensitive to some factors but not to others. Changes in assay conditions that are more sensitive to the assay following transfer to a new lab could have a greater impact on assay performance than those that are less sensitive to the assay.
Similarly, changes in reagents, instrument, bioanalyst, batch size and lab environment (room temperature, humidity, etc.) following method transfer can also impact assay performance in sensitivity, specificity, selectivity, linearity or reproducibility, etc. and result in assay failure. In another example where an analytical method was transferred to a CRO, repeated failures in precision of calibration standards but not that of QC samples were reported from the lab. This is very uncommon, close evaluation of assay results discovered that each replicate of calibration standard was placed 6 rows apart while replicate QC samples were placed next to each other. The observed high imprecision of replicate calibration standards is likely attributed to the difference in color development time as of assay reagents were added across the plate. After a consistent approach is employed among calibration standards, QC and unknown samples, the problem was resolved. Therefore, special attention to assay details is important for successful assay transfer from lab to lab and ensure reproducible results.
Summary
Owing to the importance of LBA in quantitation of macromolecules and the unique challenges compared to chromatographic assays, development and validation of LBA for quantitation of macromolecules has been discussed over the last two decades. Following the first American Association of Pharmaceutical Scientists (AAPS)/US Food and Drug Administration (FDA) Bioanalytical Workshop in 1990 focusing on bioanalytical method validation (BMV) for chromatographic and LBAs, subsequent workshops were held in 2000 and 2006 to continue in-depth discussions of these topics for LBAs. A number of conference reports and publications have been issued to provide recommendations for LBA method development and validation [5,10-12] and a series of publications in the AAPS journal have systematically reviewed the unique challenges of LBA in details [6-8]. They are valuable resources for both sponsor and CROs during LBA development, validation and sample analysis.
In summary, LBAs rely on unique knowledge and technical expertise as well as high-quality reagents. For successful method transfer, a highly specific and robust assay in conjunction with effective communications between sponsor and contract lab are the keys.
References
1. Global biologic market review and top ten biologics 2008. published online March 9, 2010. http://knol.google.com
2. FiercePharma, Biologics to top pharma sales by 2014, published online June 18, 2009 <http://www.fiercepharma.com>
3. Wang H.F. and Findlay J.W. Alternative and emerging methodologies in ligand-Binding Assays, In Ligand-Binding Assays: Development, validation, and implementation in the Drug Development Arena, M.N. Khan and J.W.A. Findlay, Eds., Chapter 13, pp 343-380, John Wiley & Sons, Inc., Hoboken, NJ, 2010.
4. K. J. Miller, R. R. Bowsher, A. Celniker, et al. Workshop on bioanalytical methods validation for macromolecules: Summary Report. Pharmaceutical Research, 2001, 18(9):1373-83.
5. C. T. Viswanathan, S. Bansal, B. Booth, et al. Quantitative bioanalytical methods validation and implementation: best practices for chromatographic and ligand-binding assays. Pharmaceutical Research, 2007, 24 (10):1962-73.
6. B Rup., D. O’Hara. Critical Ligand Binding Reagent Preparation/Selection: When Specificity Depends on Reagents. AAPS Journal. 2007; 9(2): E148-E155
7. J. Lee, H. Ma Specificity and Selectivity Evaluations of Ligand Binding Assay of Protein Therapeutics Against Concomitant Drugs and Related Endogenous Proteins. AAPS Journal. 2007; 9 (2): E164-E170.
8. J.W.A. Findlay, R.F. Dillard. Appropriate Calibration Curve Fitting in Ligand Binding Assays. AAPS Journal. 2007; 9(2): E260-E267
9. J.W.A. Findlay Specificity and Accuracy Data for Ligand-binding Assays for Macromolecules Should be Interpreted with Caution, AAPS J. 2008, 10 (3): 433–434.
10. V. P. Shah, K. K. Midha, S. Dighe, et al. Analytical methods validation: bioavailability, bioequivalence and pharmacokinetics studies. Pharm. Res. 1992, 9:588–92
11. V. P. Shah, K. K. Midha, J. W. A. Findlay, et al. Bioanalytical method validation—a revisit with a decade of progress. Pharm. Res. 2000, 17:1551–57
12. B. DeSilva, W. Smith, R. Weiner, et al. Recommendations for the bioanalytical method validation of ligand-binding assays to support pharmacokinetic assessments of macromolecules. Pharm. Res. 2003, 20:1885–1900
Dr. Huifen Faye Wang is currently a Director in Clinical Pharmacology at Pfizer, Emerging Markets Business Unit, and she is responsible for design and execution of clinical trials in Emerging Markets countries. Dr. Wang received her MD degree from Peking University, PhD degree in Pharmacology and Toxicology from the University of Utah and postdoctoral training in Cellular and Molecular Pharmacology at UC San Francisco. She joined Pfizer in 1998. Prior to her current role, she was the Head of Biotherapeutics Bioanalytical COE during 2005-2009 to provide bioanalytical support to Pfizer global biotherapeutics portfolio through internal resources and external collaborations.
This article was printed in the May/June 2010 issue of Pharmaceutical Outsourcing, Volume 11, Issue 3. Copyright rests with the publisher. For more information about Pharmaceutical Outsourcing and to read similar articles, visit www.pharmoutsourcing.com and subscribe for free.