Brief Summary
The current state of prefilled syringes (PFS), including reusable autoinjectors, passive retractor devices, and dual-chamber prefilled syringes for freeze-dried products, is discussed. Prefilled syringes allow customization of dose, dose accuracy, patient self-treatment, convenience, compliance, fast administration, functionality, and reliability. These devices additionally spare expensive biologics by reducing waste, avoiding secondary contamination, and decreasing human error.
The PFS must provide seal integrity, sterility, stability, and compatibility with biologics. It should avoid leachability and needle stick injuries. Prefilled syringes can also be a suitable alternative to costly or intellectual property (IP)-protected devices for biosimilars, a newlyemerging class of medicines.
The regulatory, medical practice, and other criteria to choose a plastic PFS as an alternative over a well-established glass PFS will be addressed. The impacts of quality and manufacturing aspects in selection of prefilled syringes are also highlighted. Further topics include: How much experience is there with plastics of approved products? What are risk factors of a plastic PFS in manufacturing and use? What are distinct physicochemical features? What are the risks of medication errors with a PFS with regard to dose delivery or misuse?
Discussion
The prefilled syringe improves convenience, compliance, allows home use and saves valuable drug, for instance, the biological used in treating rare disease. It is also convenient in terms of clinical supply where smaller batch sizes are desirable due to the high cost of biologics.
If a drug can be administered at a fixed dose, then prefilled syringes are also convenient in clinical trials due to accuracy and reproducibility. Risk is lowered when utilizing a prefilled syringe as opposed to withdrawing a product with undefined syringes and needles that differ enormously across investigational sites and countries. It is prudent to provide specific components to investigators and patients to minimize dosage variation, but a PFS is the best tactic. Patient compliance with the dose is reliable and the risk of failing on efficacy due to suboptimal doses is preventable.
PFS biologic products that are coupled with a device are “combination drugs”. These are regulated by the FDA's CDRH/CBER or CDRH/CDER and coordinated by the Office of Combination Drugs in the US. In the EU, the regulatory process is entirely different. There are EU alternatives of a Decentralized Procedure (DCP) influenced by the Europewide Coordination Group (CMD), with national representatives from 27 countries, or by the Centralized Procedure (CP), which is managed by the EMA and assessed by the Committee for Medicinal Products for Human Use (CHMP).
Prefilled syringes act as the primary container for drug products, and in regulatory terms constitute the immediate packaging in contact with the drug. Therefore, the applicant/sponsor must provide the FDA or EMA with data on the biologic/drug chemical, the physical and biological compatibility, and the biologic/drug stability through its shelf life. As the PFS is sterile, seal integrity testing results must also be provided.
Furthermore, the syringe system should be a clear, transparent, low extractable, low particle and sterile container-closure system. It should comply with USP/Ph Eur/JP compendial requirements, as well as standards such as ISO 11040-6 [1], which covers the barrel and piston. Compliance with validated sterilization methods in ISO 11135-1 (Ethylene Oxide), ISO 17665-1 (Moist Heat), and ISO 11137-1 (Radiation), or ISO 14937 (General Requirements), is also necessary [2-5].
Data on all of these aspects in an NDA/BLA or MAA inevitably leads to deficiency questions from the FDA or EMA.
Traditionally, glass PFSs have high quality standards and regulatory requirements, which must be upheld by plastic syringes as well. These include the lack of cosmetic defects and particulates. Regulators expect sterilization processes to cause damage to polymeric materials; for instance, the polymer can undergo chain scission which increases extractables/leachables. Shorter sterilization cycles might mean a reduced bioburden-controlled manufacturing environment.
As the PFS is a fixed product, attention must be paid to the PFS package label to avoid improper use, (which leads to reduced efficacy), and to minimize adverse drug reactions. The label and labeling of a drug product are the primary means by which practitioners and patients (depending on configuration), interact with the pharmaceutical product. The carton and container labels communicate critical information including the proprietary and established name, along with the strength, form, container quantity, expiration, and so on. The package insert labeling, (US PI or EU SmPC), communicates all information relevant to the approved uses of the drug, including the correct dosing and administration. Given the critical role that the label and labeling has in the safe use of drug products, it is not surprising that they comprise 33% of medication errors reported to the USP-ISMP Medication Error [6].
Being a new class of product, biosimilars represent a new paradigm. The FDA allows a non-US reference product, but also a different “presentation”, or “formulation”, of the biosimilar itself [7-9].
For instance, the FDA allows a biosimilar, “a pre-filled syringe or in an auto-injector device” (which are considered the same “injectable” dosage form), even if the reference product is licensed in a vial presentation [7]. This is an important provision in the US regulation as it indicates the FDA’s preference, namely a PFS to a vial. In addition, it addresses the possibility for a biosimilar to have a PFS presentation in cases where the originator has a biologic/drug plus device with patent or trademark protection.
Table 1. Attributes of Glass PFS

Increasing Sophistication and Market Trends
A sophisticated form of delivery includes a PFS that is mounted onto a device with an activation button, legible viewing window, safety sleeve, retractable needle, and an interlock that prevents operation until the device is firmly placed against the injection site. Naturally, this is worth the investment for a product with high projected sales and a long lifecycle. Break loose and gliding forces upon siliconization are important and can depend on flange length, and external diameter.
Oftentimes, biopharmaceutics are not sufficiently stable when formulated in aqueous solution and lyophilization is required. For this purpose, PFS dual-chamber systems (DCS) can provide safety, convenience and compliance. The main fields of application of DCS are self-administered products, emergency-drug, cytotoxic drugs, and administration of biologics such as incompatible liquids and vaccines (antigen + adjuvant).
The reconstitution procedure requires a push-turn-push plunger to separate reconstitution in a closed chamber (no contact to needle at this point) following a degassing procedure. The front stopper rests in the front hub and reconstituted drug flows through micro channels to the pre-attached needle. Baked-on silicone can be used, which enhances the durability of the coating for lower and enables more consistent friction forces, and can improve compatibility to biopharmaceutics.
As the diluent cannot be heat-sterilized in the presence of the lyophilizate, the diluent needs to be filled first and lyophilization of the product conducted in the presence of the diluent. Bubble-free filling of diluent is mandatory. Freeze-drying can be performed in the presence of the diluent stepwise. Therefore, vacuum degassing of the diluent comes first, then positioning of the middle stopper, and finally, autoclaving and lyophilization.
The rheumatoid arthritis market is an example of a maturing competitive field with many injectable devices. There are six drugs in the US that are all administered subcutaneously. The multiple sclerosis market maintains five PFS products.
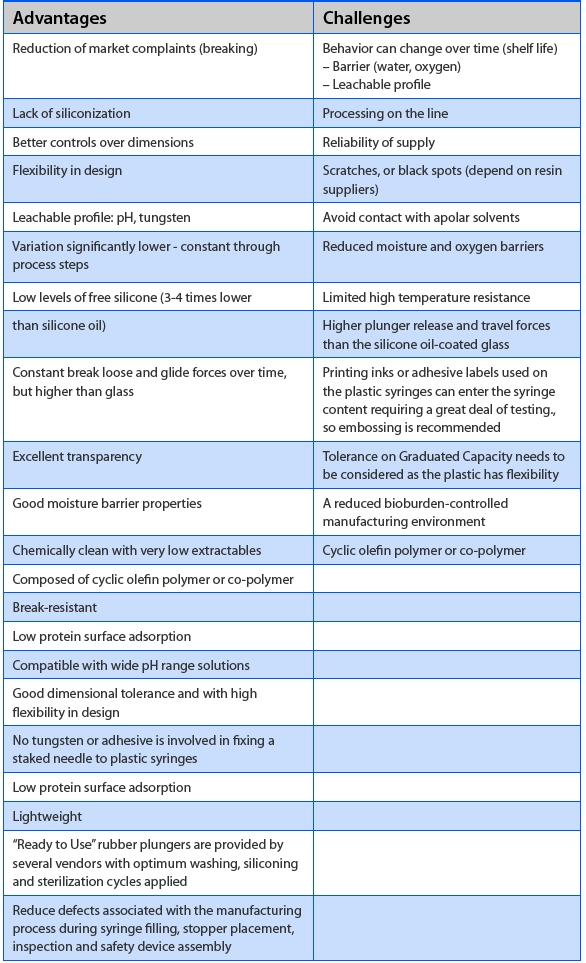
Table 2. Advantages and Challenges of Plastic PFS
Currently, many monoclonal antibodies (mAbs) administered intravenously are being developed in a second generation for subcutaneous administration, partly for patent extension to protect against biosimilars. Generally, they are infrequently administered (once per week or less). As they can be given in high doses, this requires administration of high viscosity and/or volume. Viscosities can be up to ~30 cps with administered volumes > 1 ml. The number of injections or the dosing frequency can be reduced as a result. This may call for increased needle size or new technology design.
Market expectations are based on existing technologies with low volume and low viscosity products such as:
- Movement towards smaller needle sizes (e.g. rheumatoid arthritis, multiple sclerosis)
- Auto-injector technologies
- Single-use disposables
- Higher doses administered (volume and viscosity)
- Improved FDA-based “human factors” criteria optimization
Rapid growth of injection devices in the industry has led to key challenges with regulatory expectations when changing the mode of application. FDA approval delays can take up to a year for complete response letters and requirements for clinical testing (for example, when making changes to combination products). FDA or EMA expectations differ with additional bridging testing such as bioequivalency. Also, the FDA requests “ease-of-use”, “clinical handling”, and “actual use” clinical studies with proof of adequacy of “human factors” and design validation data.
“Human factors” testing is used to inform and optimize device design concerning cognitive and ergonomic interactions with the operation of the device. It also assesses attributes of the user population and impact of the use environment, identifies and mitigates use errors in the design or instructions for use, and ensures that users can operate devices safely, effectively and at acceptable risk levels. Risk and device dependency has the potential to lead to patient harm (e.g. needle stick or incomplete dosage).
Avoiding Medication Errors and Ensuring Proper Use of PFS
In 2012/2013, the EMA identified medication errors being "any unintentional error in the prescribing, dispensing, or administration of a medicinal product” and has redefined “adverse reaction” to include it in future safety signal detection [10]. The EMA emphasized concern that the FDA has attributed 7,000 deaths annually to medication errors in the US alone. Along with the technical aspects of the PFS or related device, proper use is an important consideration. Failure Mode Effects Analysis (FMEA) uses a systematic approach with specific criteria to review container labels and carton labeling. It can determine how to design or how to change a label to meet these criteria. FMEA provides an evaluation of potential failure modes for processes and their likely impact on outcomes and/or product performance. Once failure modes are established, risk reduction can be applied to eliminate, contain, decrease or control the potential failures. FMEA relies on product and process understanding. FMEA methodically breaks down the analysis of complex processes into manageable steps. It is a powerful tool for summarizing the important modes of failure, factors causing these failures, and the effects of these failures. Mistakes can occur anywhere in the medication-use system, from prescribing to administering a drug in a variety of settings (hospitals, outpatient clinics, nursing homes, homecare, etc.).
Failure Mode Effects Analysis (FMEA) for evaluation of container labels and carton labeling reduces the risk of medication errors using the following criteria:
- Does the label clearly state its contents?
- Will this label be confused with any of the manufacturer’s other products?
- Will medical practitioners be involved?
The FDA’s Division of Metabolism and Endocrinology Products (DMEP) uses FMEA for proprietary name verification during NDA review. This is important for injectable drugs that are expensive, used infrequently, and fulfill an unmet need. Within the FDA’s Center for Drug Evaluation and Research (CDER), the Division of Medication Error Prevention and Analysis (DMEPA) reviews medication error reports.
When applying FMEA to assess the risk of a proposed proprietary name, DMEP, with the Division of Medication Error Prevention and Analysis (DMEPA), evaluates the potential for the proposed name to be confused with another drug name. FMEA allows the Agency to identify the potential for medication errors due to similar drug names prior to approval, where actions to overcome these issues are easier and more effective than those in the post-approval phase.
In order to perform an FMEA of the proposed name and label, the Safety Evaluator must analyze the use of the product at all points in the medication use system. Because the proposed product is not yet marketed, the Safety Evaluator anticipates the use of the product in the usual practice settings by considering the clinical and product characteristics. For instance, when two syringe products look identical or similar, it is possible to mix up the products. The Safety Evaluator then analyzes the proposed proprietary name in the context of the usual practice setting and works to identify potential failure modes and the effects associated with the failure modes.
The FDA’s Guidance for Industry Development and Use of Risk Minimization Action Plans (RiskMAPs), CDER/CBER says that if RiskMAPs use multiple tools or interventions, it may be useful to consider using evaluation methods applicable to the program as a whole. “For example, a systematic program evaluation model, such as FMEA can provide a framework for evaluating the individual RiskMAP components and the relative importance of each in achieving the overall RiskMAP goal or goals” [11].
References
- International Organization for Standardization ISO 11040-6:2012. “Prefilled Syringes – Part 6: Barrels for Injectables”.
- International Organization for Standardization ISO 11135-1:2007. “Sterilization of health care products – Ethylene oxide – Part 1: Requirements for development, validation and routine control of a sterilization process for medical devices”.
- International Organization for Standardization ISO 17665-1:2006. “Sterilization of health care products – Moist heat – Part 1: Requirements for the development, validation and routine control of a sterilization process for medical devices”.
- International Organization for Standardization ISO 11137-1:2006. “Sterilization of health care products – Radiation – Part 1: Requirements for development, validation and routine control of a sterilization process for medical devices”.
- International Organization for Standardization ISO 14937:2009. “Sterilization of health care products – General requirements for characterization of a sterilizing agent and the development, validation and routine control of a sterilization process for medical devices”.
- FDA, Draft Guidance for Industry Safety Considerations for Container Labels and Carton Labeling Design to Minimize Medication Errors. April 2013.
- FDA, Draft Guidance for Industry, Quality Considerations in Demonstrating Biosimilarity to a Reference Protein Product (issued jointly by CDER and CBER). February 2012.
- FDA, Draft Guidance for Industry, Biosimilars: Questions and Answers Regarding Implementation of the Biologics Price Competition and Innovation Act of 2009 (issued jointly by CDER and CBER). February 2012.
- FDA, Draft Guidance for Industry on Scientific Considerations in Demonstrating Biosimilarity to a Reference Product (issued jointly by CDER and CBER). February 2012.
- EMA, Medication-errors Workshop. Workshop Report 28 February – 1 March 2013. European Medicines Agency, London, United Kingdom
- FDA, Draft Guidance for Industry Development and Use of Risk Minimization Action Plans, March 2005. http://www.fda.gov/downloads/ RegulatoryInformation/guidances/ucm126830.pdf
- Pre-Filled Syringes & Injector Devices for Biologicals, Informa Life Sciences, Berlin, 5-6 December 2012.
Dr. Hoss A Dowlat provides strategic international regulatory support and due diligence to the pharmaceutical industry and financial institutions. Dr. Dowlat is a well-established international trainer and presenter in EU/US regulatory affairs, scientific advice, the Common Technical Dossier (MAA/ NDA/BLA), labeling (prescriber and patient), clinical safety, pharmaceutical development, combination drugs, biologics, and biosimilars. He has more than 31 years of drug development experience in over 15 therapeutic areas in the European and North American pharmaceutical industries, 21 years of which have been in regulatory affairs. He is located in Freiburg, Germany and can be reached at: [email protected]