Abstract
Pulsatile drug delivery releases drug substance at the intended time, the intended location, and in the intended amounts. This type of drug delivery system is particularly beneficial to patients suffering from chronic problems such as arthritis, asthma, hypertension and also for drugs that work as substrate to p-glycoprotein (pgp) and hence show poor permeability. These drug delivery systems are designed according to the circadian rhythm of the body. The drug is released rapidly and completely as a pulse after a lag time and these systems follow the sigmoidal release profile characterized by a time period [1].
In this research, a multiparticulate drug delivery system for a poorly permeable and poorly stable drug using combination of pulsatile releases is used. The final dosage form consists of six bead populations in a specified ratio so that six release bursts of API would occur at six different locations in the gastro-intestinal tract. This approach helps to ensure that the supersaturation of API in the intestinal tract results in passive diffusion. Therefore overcoming pgp action and achieving the desired concentration versus time profile of API in blood plasma with reduced Cmax and increased Tmax, relative to a single immediate-release dose.
Introduction
Pharmaceutical research increasingly focuses on the delivery systems that enhance desirable therapeutic objectives while minimizing side effects. In recent pharmaceutical applications involving pulsatile delivery, multiparticulate dosage forms gain favor over monolithic forms due to their potential benefits. These may include predictable gastric emptying, lowest risk of dose dumping, flexible release patterns and increased bioavailability with minimum inter- and intra-subject variability. Achieving a prolonged release in formulation development of pharmaceutical drugs that degrade with prolonged contact with gastro-intestinal fluids has always been a challenge. If such drugs act as a pgp substrate, then the formulation development becomes even more complicated as poor permeability makes it more difficult to maintain the required blood plasma level of the drug for a longer period of time [2]. In order to overcome these issues, other techniques of drug delivery such as the drug transport through paracellular or active transport could be engineered. However, such drug delivery systems can prove to be very expensive to develop and manufacture as a commercial product [3-5]. In such cases, multiparticulate systems could be used effectively to overcome p-glycoprotein expression by dose dumping [6].
Technically, pulsatile drug delivery systems administered via the oral route could be divided into two distinct types, the time controlled delivery systems and the site-specific delivery systems, thus providing special and temporal delivery [1, 5]. Pulsatile drug delivery systems can be controlled either by time or by the in-vivo environment such as pH, enzymes, gastrointestinal motility, etc. The principle rationale for the use of pulsatile release is for situations where a constant drug release, i.e., a zero-order release, is not desired. Many body functions follow circadian rhythm, i.e., their activity increases or decreases with time. A number of hormones including rennin, aldosterone, and cortisol show daily as well as hourly fluctuations in their blood levels. Circadian effects are also observed in the case of pH and acid secretion in stomach, gastric emptying, and gastro-intestinal blood transfusion [7, 8]. Many circadian-dependent diseases display symptoms in the early morning hours prior to awakening or in the morning after awakening. It is well known that patients with asthma experience symptoms at night. Dyspnoea and Peak of Expiratory Flow (PEF) values have been found to become worse during the night. Many asthma attacks occur between 4 and 6 AM. Nocturnal asthma is a complex interaction of several coincident circadian rhythms e.g. hydrocortisone and adrenalin secretion. Symptoms of allergy, e.g. runny nose, stuffy nose, wheezing and sneezing are also most frequent in the morning before breakfast [9, 10]. Ischemic heart diseases, such as angina, myocardial infarctions and strokes manifest much more frequently from 9 and 11 AM than any other time of the day or night. These systems are beneficial for drugs with chronopharmacological behavior, where nocturnal dosing is required, and for drugs that show a first-pass effect. Drugs like salbutamol sulphate produce biological tolerance and hence demand a system that will prevent their continuous presence at the site of action as this tends to reduce their therapeutic effect [11]. In such cases, pulsatile drug delivery with delayed release is highly preferred since the drug can be administered to the patient in advance. Also, if the symptoms arise at an odd time, the patient would not have to rush to find the medication.
Methodologies for Pulsatile Drug Delivery Systems (PDDS) can be broadly classified into five classes:
- Time controlled pulsatile release: a single unit system or multiparticulate system.
- Stimuli induced pulsatile release: thermo-responsive pulsatile system or chemical stimuli induced pulsatile systems.
- External stimuli pulsatile release: electro responsive pulsatile system or magnetically induced pulsatile system.
- Pulsatile release systems for vaccine and hormone products.
- Pulsatile release system which relies on colonic bacteria for release. In these systems, colonic bacteria are utilized to degrade the substrate and achieve a burst of drug substance [12-15].
The pharmaceutical drug used in this study worked as a pgp substrate and hence the permeability of the drug was very poor (BCS class III). The tablet with controlled release formulation could not provide the required drug plasma concentration over a long period of time. Therefore, a pulsatile release approach was used. The hypothesis was that with pulsatile release profile, the drug would create a supersaturation, helping the drug permeation through passive diffusion. The drug also degrades with prolonged exposure to gastro-intestinal fluids. Hence, a multiparticulate drug delivery system with multiple pulses was used to achieve the drug plasma level for a prolonged period of time. Multiparticulate-based drug delivery systems provide several advances such as more predictable gastric emptying, less susceptibility to dose dumping, and ease of manufacture when multiple pulsatile releases are required.
Extrusion spheronization was used to produce drug-loaded beads approximately 0.8 mm in diameter. The beads were split into six sub-lots and each individual sub-lot was coated with different delayed release polymers to produce either pH-dependent or pH- and time-dependent pulsatile releases. The six sub lots were then combined in a single capsule in a pre-defined ratio to obtain final dose.
Materials and Methods
Formulation development
Formulation development was conducted by selecting and optimizing the material composition to provide pH-dependent pulsatile releases at labscale. Extrusion spheronization was used to produce core beads with 85% w/w drug loading. Because microcrystaline cellulose (15% w/w) works as an extrusion and spheronization aid, this was used as an excipient. The drug and microcrystalline cellulose were granulated with water to produce a wet mass. The wet granulation was then extruded through a Luwa extruder with a 0.8 mm screen to produce noodle type extrudates. The extrudates were then spheronized using a spheronizer equipped with a 2 mm cross hatch plate.
To obtain spherical beads, the formulation and process parameters for extrusion and spheronization were optimized. The resulting beads were spherical in shape and had a narrow particle size distribution of approximately 0.8 mm. The beads after spheronization were dried using a fluid bed dryer to achieve the moisture target of less than 3% w/w in the beads as measured by loss in drying. The dried beads were sieved to remove any agglomerates and fines and then divided into six smaller sub-lots for coating studies. Table 1 shows the process parameters used to produce the drug-loaded core beads.
Table 1. Manufacturing Process Details
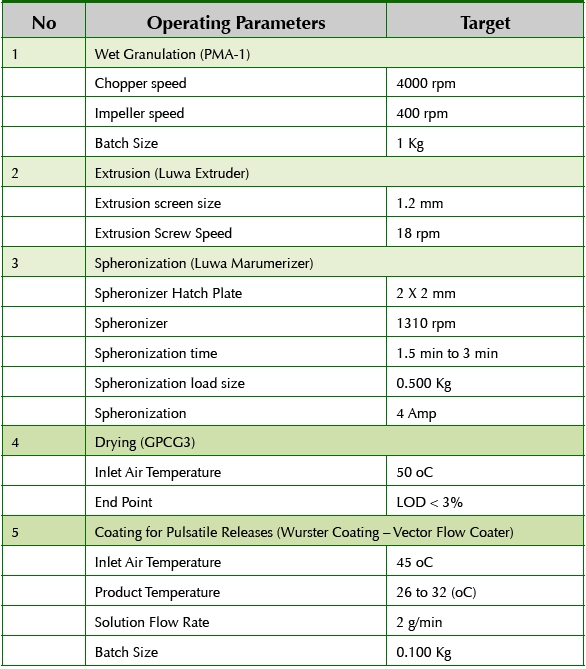
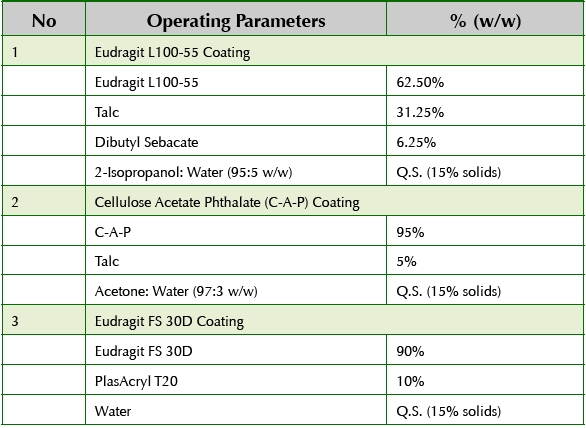
Sub-lots of core beads were coated with specific polymers in order to create six bead populations with unique pH-dependent or pH- and timedependant pulsatile releases. Table 2 shows the details of the coating suspensions prepared to produce the various coated bead populations.
Table 2. Pulsatile Bead Formulation Details
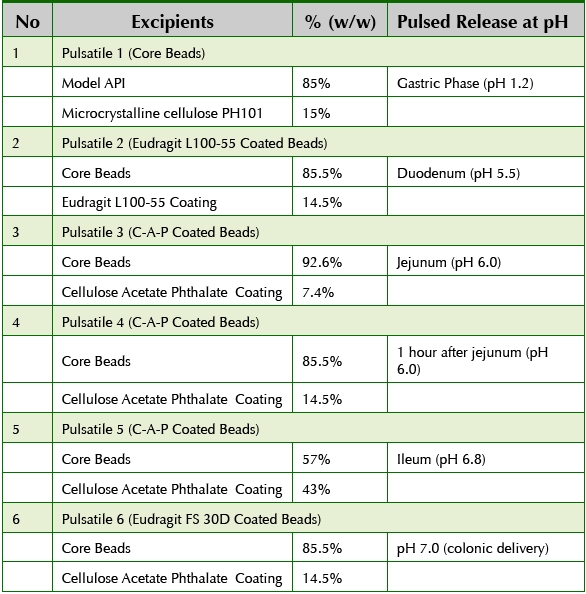
The first pulsatile release was obtained from core beads which release the entire dose in gastric fluids in less than 15 minutes. Due to very rapid releasing formulations, the core beads were ideal to use as substrate to produce the remaining five bead populations since according to FDA guidance, matching F2 for core beads of different bead populations were exempted. The second bead population was achieved by coating core beads with Eudragit L100-55 which produced the API release in the duodenum (pH around 5.5). Cellulose Acetate Phthlate (CAP) coating was applied to core beads to produce third bead population which produced an API release pulse in the jejunum (ph around 6.0). For the fourth population of beads, CAP coating was applied to a higher weight gain so that the pulsatile release of API could be obtained about 1 hour after the fourth bead population reaches jejunum. For the fifth bead population, the CAP coating was applied to an even higher weight gain so that the API release was achieved in the ileum (pH around 6.8). For the sixth bead population, core beads were coated with Eudragit FS30D to obtain a pulsatile release in the colon (pH around 7.0). Once all six populations were manufactured, the beads were blended using V-blender and then filled in size 0E capsules to produce the final dosage form. The proportion of each bead population was optimized empirically by conducting several in vitro dissolution tests.
Dissolution method development
Various dissolution test methods were used as a part of analytical and formulation development in order to achieve a discriminating dissolution method. The two-step sequential dissolution (2 hours in pH 1.2 followed by 10 hours in pH 6.8) did not show enough discrimination in API release from the five bead populations. It was concluded that the sequential dissolution using USP APP I at 100 rpm (2 hours in pH 1.2 followed by 2 hours in pH 5.5, followed by 2 hours in pH 6.0, followed by 6 hours in pH 7.4) was a satisfactory method to produce the discrimination in dissolution profiles and was used to assess the formulations for the comparative pharmacokinetic study.
Results and Discussion
The in vitro dissolution results showed that the core beads displayed a pulsatile release of drug in 15 minutes as soon as they reached the simulated gastric fluids. The remaining five bead populations were designed to have a pulsatile release from the duodenum to the colonic region. Hence, the API was delivered throughout the gastro-intestinal tract from the gastric region to the colon in the form of 6 pulses over a period of 12 hours. By combining these six bead populations, it was possible to develop a singledose product to achieve the desired drug plasma concentration for over 12 hours, as inferred from the in vitro data. Table 3 shows the formulation details of individual bead populations.
Table 3. Pulsatile Bead Formulation Details
In order to eliminate the necessity of implementing the similarity factor (F2) for core beads, the desired dissolution profile of cores was established as at least 85% API release in less than 15 minutes. The core beads released 100% in less than 15 minutes so they were ideal as a substrate to manufacture the rest of the five bead populations. The optimized dissolution profiles for the six bead populations are shown in Figure 1. The dissolution profiles show enough discrimination based on pH/time to theoretically obtain six distinct pulsatile releases in-vivo. Figure2 shows the composite dissolution profi le of the six bead populations fi lled in the size 0 capsule. The composite dissolution profi le suggests pseudo zero order release of the drug over 12 hours.
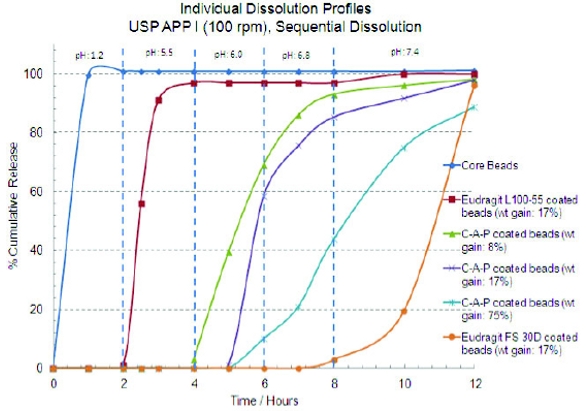 Figure 1. Pulsatile release profiles of individual bead populations
Figure 1. Pulsatile release profiles of individual bead populations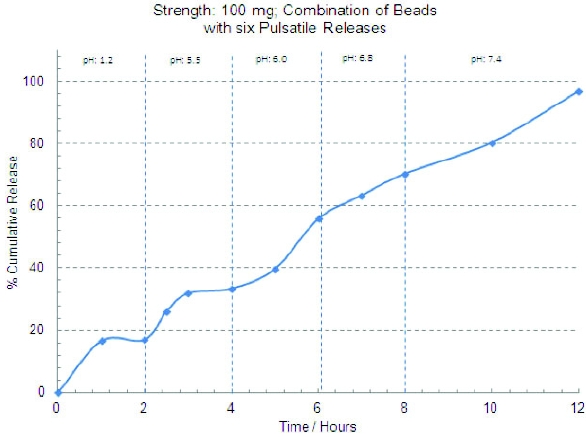 Figure 2. Dissolution profi le of the combined bead populations
Figure 2. Dissolution profi le of the combined bead populationsConclusion
A formulation consisting of six bead populations with six distinct pulsatile releases was successfully developed. USP APP I (basket) dissolution method with sequential dissolution at 100 rpm was found to be a discriminating method to develop the product. The friability of core beads was found to be satisfactory (< 1.0%). This approach helped to ensure that the concentrations of API in the intestinal tract resulted in the desired concentration versus time profi le of API in blood plasma, with reduced Cmax and increased Tmax, relative to a single immediate-release dose. By combining six populations of beads, it was possible to develop a singledose product to achieve the desired drug plasma concentration for over 12 hours, as inferred from the in vitro data.
This approach was found to be a suitable alternative to self-emulsifying drug delivery systems which are typically used as the preferred drug delivery platform to formulate BCS class III compounds. As the drug is poorly stable in the gastro-intestinal fl uids, the pulsatile release approach was found to be useful since the drug in the individual bead populations had limited time contact with the GI fl uids. With combining multiple bead populations, the composite release remained near zero order release as inferred from the in vitro data.
References
- Pallab Roya, Aliasgar Shahiwala, Journal of Controlled Release, 134 (2), 74-80 (2009).
- Hans Maag, Drug Discovery Today: Technologies, 9 (2), 121-130 (2012).
- Ryo Yoshida, Kiyotaka Sakai, Teruo Okano, Yasuhisa Sakurai, Advanced Drug Delivery Reviews, 11, 85-108 (1993).
- V.S. Mastiholimath, P.M. Dandagi, S. Samata Jain, A.P. Gadad, A.R. Kulkarni, International Journal of Pharmaceutics, 328, 49-56 (2007).
- Alessandra Maroni, Lucia Zema, Maria Dorly Del Curto, Giulia Loreti, Andrea Gazzaniga, International Journal of Pharmaceutics, 398, 1-8 (2010).
- Joseph Kost, Robert Langer, Advanced Drug Delivery Reviews, 6, 19-50 (1991).
- Joao F. Pinto, International Journal of Pharmaceutics, 395, 44-52 (2010).
- Bruce J. Aungst, Hiroshi Saitoh, Deborah L. Burcham, Shiew-Mei Huang, Shaker A. Mousa, Munir A. Hussain, Journal of Controlled Release, 41 (1-2) 19-31 (1996).
- Sam Maher, David J. Brayden, Drug Discovery Today: Technologies, 9 (2), 113-119 (2012).
- Virginia Zabaleta, Gilles Ponchel, Hesham Salmana, Maite Agüeros, Christine Vauthier, Juan M. Irache, European Journal of Pharmaceutics and Biopharmaceutics, 81 (3), 514-523 (2012).
- Alessandra Maroni, Lucia Zema, Maria Dorly Del Curto, Giulia Loreti, Andrea Gazzaniga, International Journal of Pharmaceutics, 398 (1-2), 1-8 (2010).
- Georg Brabant, Klaus Prank, Christoph Schofl , Trends in Endocrinology and Metabolism, 3 (5), 183–190 (1992).
- Alfredo Martínez, Jaqueline Arias, Jorge A. Bassuk, Heng Wu, Paul Kurlansky, Jose A. Adams, Peptides, 29 (1), 73–78 (2008).
- James D. Sink, W. Randolph Chitwood Jr, Ronald C. Hill, Andrew S. Wechsler, The Annals of Thoracic Surgery, 29 (1), 57–62 (1980).
- L. Maggi, U. Conte, P. Giunchedi, P. Colombo, International Journal of Pharmaceutics, 99 (2–3), 173–179 (1993).
Dr. Abhijit Gokhale is a Senior Scientist (Formulations and Process Development) at Patheon Pharmaceuticals (since May 2011). Dr. Gokhale has over 12 years of drug development experience in both academic and industrial environments. He has been intricately involved in formulating pharmaceutical drugs for both controlled release as well as bioavailability improvement applications.
Dr. Thomas Williams is currently a Manager of Formulation and Process Development at Patheon Pharmaceuticals. Dr. Williams has 15 years of drug development experience, with the last 7 years spent at Patheon. He has extensive experience in formulation development, process development, and scale-up to commercial manufacturing.
Dr. Jason Vaughn is a Director, Formulation and Product Development at Patheon Pharmaceuticals. Dr. Vaughn has over 13 years of experience in the pharmaceutical industry. His expertise includes; formulation development of solid oral dosage forms, liquids and semisolids, solubility and bioavailability enhancement, and early and preclinical development