The pharmaceutical industry is inherently risky. To develop a molecule that has a desired, efficacious but yet safe effect in humans is a remarkable achievement considering the numbers. If all the molecules in discovery and development were added together over recent years and compared to the number of successful drug approvals each year, the ratio is (approximately) 10,000:1.1 In other words, for every 10,000 molecules entering drug discovery, 9,999 will fail during some stage of the development process and only one will become a successful marketed drug (and even then, it is not certain development costs will be recouped). Couple this extraordinarily high attrition rate with a lengthy, complex and expensive process (average 12 years in development and cost of £1.15 billion1) and it is clear that drug development is an industry where the risk of expensive failure is high. So, is it possible to turn these odds a little more in our favor? Can we better select new molecules that will have a greater than a 1:10,000 chance of progressing though development and making a successful, safe, marketed product? One of the keys to answering this forms the focus of this paper. By better understanding the risk areas where molecules fail and why, and by applying appropriate experimental resources to identify potential exposure to these risks early in the development process, we will help identify molecules that have a better chance of survival. This is the basis of ‘de-risking’.
Advancements in in vitro technologies and analytical instrumentation over the past decade allows scientists to apply a variety of in vitro based assays designed to rapidly assess risk-liability to some of the major factors contributing to both current high attrition rates and also delays in development. in vitro assays are ideal in early phase development they typically can be conducted quickly and involve low compound usage – requiring just milligrams of material. This makes in-vitro assays ideal for early discovery programs, where typically, only small amounts of compound are available. These specific in vitro assays can be efficiently combined into a “risk assessment package” which can be employed early in drug development as a de-risking paradigm to aid informed decision-making prior to major investment decisions on new drug candidates.
The risk-liabilities under focus in this paper are metabolically-driven and comprise just one area of potential attrition risk (another major risk area, beyond the scope of this paper, is lack of clinical efficacy reflected by failure to transit from Phase II to Phase III during clinical development). Regarding metabolic risk then, one of the most important and yet sometimes under-appreciated parameters of drug development is drug metabolism. To fully understand how a new molecule is metabolized in the body is crucial to successful drug development. Drugs are metabolized by families of drug metabolizing enzymes (DMEs) which are primarily, but not exclusively, located in the liver. The most important of these is cytochrome P450 (often referred to by its root gene name of CYP). Collectively the various sub-families of CYP enzymes (primarily CYP1A, 2A, 2B, 2C, 2D, 2E, 3A) currently metabolize approximately 70% of all prescribed small molecule medications.2 Consequently, drug-CYP interactions are a major mechanism of [pharmacokinetic-based] drug-drug interactions between co-administered medications in humans. For small molecule drugs, drug metabolism will often drive clearance (metabolism is the major clearance mechanism for neutral, lipophilic small molecule drugs), pharmacokinetics (including population exposures in poor and rapid metabolizer sub-groups), drug-drug interaction potential and some forms of toxicity.
An adverse liability in any of these parameters is highly undesirable, causing approval delays at best to potentially catastrophic post-market withdrawal at worst as illustrated in Table 1.
Table 1. Potential major development risks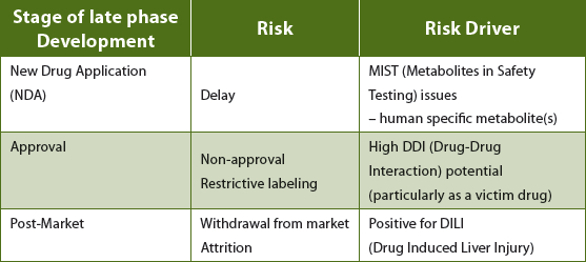
The de-risking strategies suggested in this paper are designed to investigate if a molecule may have a liability in these risk areas, based upon how it is metabolized and how it interacts with CYP enzymes (and drug transporters). If a molecule is tested, and is low or negative in these in vitro assays, it can be considered somewhat ‘de-risked’ from these metabolically-driven potential problem areas. If a molecule is positive in these tests, then the developing company is now risk-aware and this knowledge can be used to better assess risk versus benefit before deciding whether to continue to progress the molecule or not.
The individual in vitro assays applied in a de-risking strategy, are (sometimes) not by themselves, entirely predictive and the chance of a false negative always exists if a single assay approach is used. For example, a false negative compound would include a drug that causes drug induced liver injury, but is not necessarily associated with a high level of covalent binding in human liver microsomes. However, very often a “red flag” will be somewhere, and will usually be found by applying a matrix of in vitro tests, making the metabolic de-risking program a powerful tool to detect major risk alerts.
If liability is detected early enough, it may be possible to make changes to the structural chemistry of the discovery program, which retains the pharmacophore, but reduces the metabolic liability by blocking certain metabolic pathways and preventing particular [reactive/toxic] metabolites from forming (the early volatile anesthetics and the Type II diabetes insulin sensitizer glitazone class of drugs are examples of this approach).
Ultimately of course, it is the aim of the pharma industry to develop medicines, which are efficacious, but also safe to use in humans. However, in addition to purely the safety aspect, a natural by-product of de-risking is that a de-risked molecule, when compared to a similar but non-de-risked molecule (all other things being equal), is intrinsically more valuable. A de-risked molecule will aid in/out-licensing (duediligence) and ultimately provides an increased measure of scientific confidence to invest.
The primary aim of an early de-risking strategy is to provide a comprehensive and rigorous data set, which allows a rational, scientifically-based assessment to be made about the ‘likelihood’ that a molecule may carry a liability, before major investment decisions are made. With this in mind, integrating metabolic de-risking into drug discovery would seem to be an advisable strategy given that the pharma industry operates in an increasingly risk-adverse environment.3
The three main elements of metabolic de-risking are discussed below.
Drug Induced Liver Injury
Drug-induced liver injury is a major concern for the pharmaceutical industry. It is the single biggest cause of liver transplant in humans and a major cause of safety-related drug withdrawal post-market. It is also a major cause of drug attrition during development.
DILI can be of both direct Type A toxicity – which is dose-dependent and reproducible and is therefore predictable (such as with acetaminophen/ paracetamol), or it can be Type B, often referred to as idiosyncratic, which is not generally predictable. This type of toxicity is particularly problematic as it is generally not dose-dependent (although there does appear to be some relationship with total daily dose) and is not predicted from animal safety studies. Idiosyncratic DILI is generally a rare event – and is characterized by low frequency (as few as 1:10,000 individuals may be affected). However, where it does occur its effect can be extremely serious and associated with high morbidity. Its low frequency means that often it is not detected during clinical trials. Hence detection often occurs once the drug is approved and moves into a much larger patient population, sometimes becoming a major reason for post-market withdrawal).
The decision to progress a molecule that is displaying DILI-positive characteristics is an extremely high risk decision and should only be made after taking into account multiple factors including the therapeutic area, whether the molecule will be meeting a currently unmet need (first in class) and likely human dose (lower human doses significantly reduce the risk of DILI). Potential impact scenarios are outlined in Table 2.
Table 2. Potential impact of DILI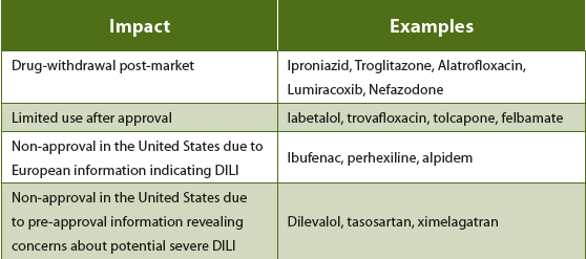
Although precise mechanisms of DILI are still being understood (including mitochrondrial dysfunction and inhibition of drug transporters for example), one pathway increasingly believed to contribute is via the formation of reactive metabolites (RM). RMs are short-lived, highly reactive, electrophilic (+ve) metabolites (sometimes radicals) which can bind to macromolecules such as hepatic proteins, antibodies and DNA causing both Type A (direct, e.g. genotoxicities) and Type B (idiosyncratic, e.g. DILI) toxicity. If the binding is covalent (irreversible) then this can trigger immune related, hapten-like anaphylactic shock responses resulting in tissue/organ necrosis. Notable examples of a positive correlation between RM formation and clinical toxicity include clozapine, troglitazone, diclofenac and nefazodone (among many others).
The detection of RM formation is therefore a key component of derisking strategy. Today, RM formation can be assessed using various in vitro assays, either directly using so-called RM trapping assays or via surrogate markers such as covalent binding or time-dependent inhibition of CYP enzymes.
Covalent Binding
In these assays, the molecule is incubated with human liver microsomes and following a period of metabolic bio-activation the samples are exhaustively extracted to recover all of the drug-related material. A shortfall in mass-balance (inability to be able to extract all total drugrelated material) can be indicative that the test compound is being metabolically activated to a metabolite, which is covalently bound to the microsomal protein. Positive and negative controls should always be included to assess how the level of binding with the test compound compares to known drugs that are DILI-positive and those that are not covalently bound and are negative for DILI. For this assay, having a radiolabelled form of the drug to use is preferable.
Reactive Metabolite Trapping
In these assays, the drug is again incubated with human liver microsomes, but this time in the presence of an excess of a strong nucleophile, often glutathione (GSH). Any electrophilic (reactive) metabolites produced are ‘trapped’ in situ by reaction with GSH. Samples are analyzed by LCMS/MS using neutral loss (m/z 129), full scan, product ion or precursor scan techniques as appropriate to assess the number, and if required, the structure of any reactive metabolites produced. As with covalent binding, appropriate controls are included (compounds which are positive and negative for both reactive metabolite formation and clinically relevant DILI).
Covalent binding and RM trapping comprise two very useful assays for DILI risk assessment. However, ideally, as alluded to previously, the overall assessment should not be made on one or two parameters alone. Other crucial aspects to consider when making the overall DILI risk assessment are potential inhibition of BSEP transporter (organ-specific toxicity),4 structural alerts (e.g. potential acyl glucuronide or quinone type metabolite formation),5 lipophilicity (risk is elevated if log>P3)6 and human daily dose (risk is elevated if>100mg/day).)6
If the test drug is low/negative for the DILI characteristics outlined, then it is [likely] considerably de-risked from major safety-related issues including attrition and potentially catastrophic drug withdrawal post-market.
The overall DILI de-risking strategy used at Envigo is presented in Figure 1.
 Figure 1. DILI de-risking strategy
Figure 1. DILI de-risking strategyDrug-Drug Interactions (DDI)
The assessment of pharmacokinetic-based drug-drug interactions (DDI) is a major element of modern drug development. As populations expand and individuals live longer, the occurrence of patients being prescribed multiple concurrent medications (polypharmacy) is increasing.7 Consequently the risk of adverse drug reactions due to DDIs is also increasing and this area of drug development is coming under increasing regulatory scrutiny. Drugs with a high DDI potential (particularly as a victim drug) may be at risk of non-approval, excessive labeling or post-market withdrawal.
To test a new drug for DDI potential, regulatory authorities recommend the initial use of a range of in vitro studies (basic models) designed to assess interactions of new drugs as both a victim and perpetrator of DDIs:1,2,3
- Perpetrator drug studies – to assess how the test drug will affect other medications – these studies assess the potential inhibition and induction of important drug metabolizing enzymes such as CYP and UGT and transporter proteins by the test drug.
- Victim drug studies – to assess how the test drug will be affected by other medications – these studies determine which enzymes and transporter proteins are involved in the clearance of the test drug (substrate/phenotyping interactions).
A detailed review of each of these assays is beyond the scope of this publication, however it is important to appreciate the risk associated with each category. Often, it is assumed that the bigger risks are as a perpetrator drug – these are drugs that cause pharmacokinetic changes in other co-administered medicines. Generally however, for drug approval, this is an incorrect assumption. Perpetrator interactions such as [reversible] CYP inhibition or CYP induction can be tolerated to a certain extent by dose adjustments and/or labeling. The bigger risk lies as a victim drug.
If a drug has a single enzyme (or transporter) dominating its clearance then this will be of regulatory concern, unless the drug has an established wide therapeutic window. The dominance of just a single enzyme or transporter to the clearance of a drug is referred to as Fraction Metabolized (Fm) or Fraction Transported (Ft), respectively. Ideally, for individual enzymes, Fm should be 0.2-0.4 as this indicates that the drug is being metabolized by multiple enzymes and hence has multiple exit pathways from the body. If, however, the metabolism of the drug is dominated by a single enzyme, so Fm approaches 0.8-1.0, then it is extremely vulnerable to the effects of co-administered drugs which may be inhibitors or inducers of that particular enzyme (causing drug accumulation of the victim drug or increased clearance, respectively). In cases where the Fm (or Ft) is high (>0.8), drug-drug interactions are greatly magnified,8 this is a major concern with single-substrate drugs and without a wide therapeutic window, the drug may be heavily labeled (reducing competitiveness), not be approved (e.g. debrisoquine, perhexilline) or even withdrawn from the market (e.g. terfenadine, cerivastatin, astemizole, cisapride)
The overall DDI de-risking strategy used at Envigo is presented in Figure 2.
 Figure 2. DDI de-risking strategy
Figure 2. DDI de-risking strategyMetabolites In Safety Testing (MIST)
Over the past two decades considerable industry and regulatory discussion has focused around the importance of drug metabolites as potential sources of drug toxicity. In 2008, the FDA published draft guidance on this issue entitled “Safety Testing Of Metabolites"9 with an updated guidance issued just recently in 2016.10 Collectively, the issue of metabolite-mediated toxicity, and when it should be assessed, has become known as Metabolites in Safety Testing or MIST within the industry.
The principal recommendations of the FDA’s guidance regarding the concept of MIST guidance are:
- To encourage the identification of differences in drug metabolism between animals used in nonclinical safety assessments and humans as early as possible during the drug development process
- To encourage the identification of differences in drug metabolism between animals used in nonclinical safety assessments and humans as early as possible during the drug development process
- Generally, metabolites identified only in human plasma or metabolites present at disproportionately higher levels in humans than in any of the animal test species should be considered for safety assessment.
- Human metabolites that can raise a safety concern are those formed at greater than 10 percent** of parent drug systemic exposure at steady state.
For any new drug in development, therefore, an early understanding that potential MIST issues (human-specific metabolism) will not arise is crucial to help ensure the metabolic relevance of the animal models used in its safety assessment (toxicity testing).
The definitive human metabolism data set is obtained from conducting a human radiolabelled (14C) AME study (Absorption, Metabolism, Excretion), where a radioactive form of a drug is given to a small number of human volunteers (n = 6-8 cohorts). However, typically this crucial study is often conducted during late phase development (after Phase II/proof of concept). This may seem a prudent approach given these human radiolabelled studies are not inexpensive to run. However, if the definitive human data set is obtained too late, it can result in significant delay(s) in the NDA/approval process should it reveal a disproportionate human metabolite to which animal models used in safety assessment were not adequately exposed.
Any [major] human-specific [disproportionate] plasma metabolites will require additional safety assessment, which in turn requires a large scale synthesis of the metabolite - a time-consuming and expensive late-stage requirement. Ultimately of course, possible termination of the drug may be warranted if toxicity is observed (with any of the metabolite(s)).
To assess MIST risk, an integrated use of both in vitro and in vivo studies should be employed, such as given in Figure 3, to inform if humanspecific metabolism could be an issue. If it is, then the human 14C AME in vivo study should be prioritized and conducted with some urgency to obtain the definitive human metabolite profile.
 Figure 3. MIST de-risking strategy
Figure 3. MIST de-risking strategyOne of the major advantages of using in vitro techniques for early de-risking is the ability to generate human metabolic data (from microsomes and/or hepatocytes) early in the development process.
Metabolic profiles can be quickly obtained using human hepatocytes and compared with animal species to investigate the formation of possible human-specific, or disproportionate, metabolites. These in vitro data can then be compared to in vivo metabolic data (plasma, blood, feces) to gain an understanding of the strength of the in vitro - in vivo extrapolation (IVIVE), which is crucial to the MIST assessment.
If after conducting a MIST strategy such as proposed above, there are no major differences between the metabolites formed in the relevant animal models in vivo and the human in vitro (hepatocyte) metabolism, then there is a scientific, rational basis for not needing to prioritize a human 14C AME study early, as the animal in vivo metabolites (and exposures) are likely to be reflective of human metabolism.
However, if there are differences between animal in vivo and human in vitro (hepatocyte) metabolite profiles, then there is risk of a MIST issue (inappropriate animal species used for safety assessment and/or human-specific metabolism) and there is therefore a need to prioritize the human 14C AME study early to obtain the definitive human metabolite data.
Summary
De-Risking is the intelligent application of early in vitro testing to investigate areas of major risk for a new molecule in development, helping to avoid spending time and resource on a compound that may carry with it a high potential liability of non-approval or market withdrawal. De-risking provides information that allows for more informed decision-making in the high-risk arena of drug development.
References
- www.pharmaceutical-journal.com/publications/tomorrows-pharmacist/drugdevelopment-the-journey-of-a-medicine-from-lab-to-shelf/20068196.article
- Clinical importance of the cytochromes P450, Nebert, Daniel W et al. The Lancet , Volume 360 , Issue 9340 , 1155 - 1162
- Lessons learned from the fate of AstraZeneca's drug pipeline: a five-dimensional framework, David Cook et al. Nature Reviews Drug Discovery 13, 419–431 (2014)
- Human drug-induced liver injury severity is highly associated with dual inhibition of liver mitochondrial function and bile salt export pump. Aleo et al. Hepatology. 2014 Sep; 60(3).
- Structural Alert/Reactive Metabolite Concept as Applied in Medicinal Chemistry to Mitigate the Risk of Idiosyncratic Drug Toxicity: A Perspective Based on the Critical Examination of Trends in the Top 200 Drugs Marketed in the United States, Antonia F. Stepan et al. Chem. Res. Toxicol., 2011, 24 (9), pp 1345–1410
- High lipophilicity and high daily dose of oral medications are associated with significant risk for drug-induced liver injury. Chen M1, Borlak J, Tong W. Hepatology. 2013 Jul; 58(1) : 388-96.
- Polypharmacy: A Global Risk Factor for Elderly People, Rushabh J Dagli and Akanksha Sharma, J Int Oral Health. 2014 Nov-Dec; 6(6).
- Guidance for Industry. Drug Interaction Studies – Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations, Draft Guidance, Center for Drug Evaluation and Research (CDER), Food and Drug Administration (FDA), February 2012.
- Guidance for Industry Safety Testing of Drug Metabolites, U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), February 2008, Pharmacology and Toxicology
- Guidance for Industry Safety Testing of Drug Metabolites, U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Revision 1, November 2016, Pharmacology and Toxicology
Guy Webber, MSc, is the Scientific Manager, In Vitro and Drug-Drug Interaction Sciences at Envigo (www.envigo.com). He manages the In Vitro Sciences Team and also future scientific strategy and services. His main research interests include developing the use of primary human cell models to better predict human DDIs, drug transporters and the role of biotransformation in drugmediated toxicity.