While GMP for clinical supply manufacture is written vaguely and “interpretable,” at least there are long established guidelines, both formal (e.g., Code of Federal Regulations, International Conference on Harmonization) and informal (e.g., Gold Sheet, Guidelines for Industry). Guidance for the use of comparative agents in the clinic, however, has until only recently been missing from those formal and informal guides, and the industry has extrapolated a wide variety of practices and procedures from the manufacturing guidelines. The philosophies and strategies used by the Comparative Agent Development (CAD) group of Pfizer Research & Development and outlined in this article are based on industry guidance, and have evolved based on years of experience with a wide variety of products, dosage forms and regulatory filings. The contents of this article reflects Pfizer’s current practices for qualification of the blinding of comparative agents and placebos. The practices will continue to evolve as the industry changes.

As recently as January 2010, in fact, the European Medicines Agency (EMEA) has released documents relating more specifically to comparative agents [1], and the European Commission updated their 2003 issue of GMP Annex 13 in February 2010 [2]. However, the Pfizer CAD group has long used two SUPAC guidance documents [3,4] to define the level of “modification” or “change” upon which the subsequent level of qualification is based. These guidelines are for “post-approval” changes and comparative agents are “post-approval” commercially available drug product. Both SUPAC guides provide the following definition: “Level 1 changes are those that are unlikely to have any detectable impact on formulation quality or performance. […] Level 2 changes are those that could have significant impact on formulation quality and performance. Level 3 changes are those that are likely to have a significant impact on formulation quality and performance" [3,4]. The Pfizer CAD group refers to these levels as negligible (level 1), minor (level 2) and major (level 3). In the case of comparative agents, the method used for blinding the commercial dosage form is the “postapproval change,” and once the level of impact of the blinding, or “change,” is determined, the amount of qualification work needed can be determined.
The EMEA guidance on documentation for Investigational Medicinal Products (IMPs) begins the definition of qualifying a blinded comparator that will be developed more fully in this article. The guide states,
"As the marketing authorisation holder (MAH) of a comparator product is only responsible for the unchanged product in its designated and authorised packaging, there is a need to ensure that the quality of the product is not negatively affected by the modifications performed by the applicant or sponsor of the clinical trial, with special emphasis on the biopharmaceutical properties. […] The modifications of the authorised comparator product should be described and their influence on the quality of the product discussed. Special focus should be assigned to all parameters relevant for the function, stability and efficacy of the medicinal product, such as in vitro-dissolution and pH-value. It should be demonstrated that these parameters remain comparable to those of the unmodified product. In case of solid oral dosage forms, comparative dissolution profiles of both original and modified comparator product should be provided to ensure unchanged bio-pharmaceutical properties. In those cases where comparability cannot be established in vitro, additional clinical data to support equivalence may be necessary” [5].
Working with these guidelines underlying their comparative agent procedures, the Pfizer CAD group advises supply, manufacturing, and clinical team colleagues when comparative agents are needed for clinical studies. These discussions always include a number of risk analyses from a variety of strategic viewpoints.
Risk Analysis – A Delicate Balance
When determining comparative agent strategy for clinical studies, a balance between Clinical Considerations, Business Considerations, and Technical Considerations is needed. A dialogue between the laboratory, supply, and clinical experts must begin early in the process, understanding that each group’s needs may oftentimes be in conflict with one another. Final decisions, then, are based on analysis of the risks in each of the areas of consideration.
Of course there are many Clinical Considerations in the decisions for comparative agent strategy of a study. The type of clinical trial will impact how “identical” the blinded Comparative Agent must be relative to the commercial product. For instance, will the dosing be done in the clinic or in the patient’s home, is the study a Single or Double Blind. Certainly the blinding method must be acceptable to the clinical team, for example, does the comparator “rattle” (an over-encapsulated tablet without backfill), look unappealing (overprints), or otherwise encourage curiosity to open (device covers) and therefore risk breakage of the blind. The purpose of the study will influence comparative agent strategy as well. Specifically, if the trial is designed to support a superiority claim, the trial needs to utilize an innovator’s product, while other studies could use generic product as the comparator. The clinician must consider the targeted population. For example, large capsules can’t be given to patients with swallowing difficulty, or product packaged in difficult blistering would be a problem for patients with arthritis. Finally, the decision regarding how “close is close enough” for blinding is dependent on how closely the subject can examine the dose or if the clinician will ever see the placebo and active together. Sometimes close is close enough. But even cost and timing are usually in the mix of clinical considerations as well – it’s not just the science.
Examination of the Business Considerations reveals a number of things to balance here as well. Both analytical qualification and manufacturing of the blinded comparator have significant timing and cost issues, typically on opposite ends of the spectrum. For instance, over-encapsulating a tablet may be the quicker, cheaper manufacture, but requires more qualification and development from the laboratory, while de-inking tablets needs very little analytical development and qualification, but is costly and time consuming to perform from the manufacturing perspective. While a deink can be done for small studies and require very little development and data, it’s too costly and impractical for a large study. So do you blind one way in the early phases and postpone the analytical spend until reaching later larger studies? It’s a decision to make. This balance of time and cost between the analytical and manufacture perspectives is achieved by answering a series of questions in a decision-tree type analysis. Again, this dialogue must be opened with the clinician early on to keep studies on track and costs managed effectively. The questions generally include:
- What is the process to blind a commercial product versus the process to formulate and manufacture a matching placebo in double blind studies?
- Is the comparator for a small early phase study or a larger, multiarm and/or global late phase study?
- Are there legal and/or regulatory issues such as trademarks, market authorization holders, country-specific requirements?
- Can the comparator be procured non-commercially marked (at times negotiated with generic suppliers)?
The business considerations, then, are mainly related to when and where the team wants to spend the resources – manufacture, or analytical qualification/early in the development, or later as trials progress.
Having touched a bit on the analytical considerations, we do need to delve deeper into these. There are a number of Technical Considerations when selecting a comparative agent and blinding strategy for a clinical study. Now the science does impact the decision making. The most obvious consideration is the Dosage Form – solid, liquid, parenteral, devices – as there are different approaches to blinding for different dosage forms. Next consider the bioavailability and speed of onset. There are different qualification requirements for different types of drug absorption – immediate release, delayed release, controlled release. When overencapsulating, excipients already common to the commercial formulation are best for backfill. If you have to use something different it is a Level 3 change according to the SUPAC documents. If a matching placebo is to be formulated, the physical properties must be considered. The team may need to match appearance (suspensions, emulsions, color, opacity), taste, texture, feeling in mouth or nose (oral liquids/suspensions, inhalants, nasal sprays), or possibly smell. Included in the technical analysis are devices or unusual primary packaging considerations. When re-packing a commercial product, or packaging a blinded comparator, it’s imperative to use equivalent or more protective packaging because the innovator’s use period and stability data would only be supported by this. Unfortunately, at times it can be difficult to determine packaging requirements, as they are not described in labeling or inserts. Blistering plastics are fairly common, but consider, for example, the material and thickness of a foil pouch that may or may not be product protective. Some other unusual primary packaging examples include dosing spoons integral with caps, eye droppers, and pre-filled syringes. These all make blinding more difficult and analytical qualification considerations more impactful. Use period is the final technical consideration as it needs stability data to support and cannot be longer than the commercially assigned expiration. Availability of supply and length of use period often influences manufacturing timing, and therefore influences comparative agent strategy.
Double Dummy Blinding is common, and with this blinding strategy, the comparator doesn’t need to match the investigational drug. Instead, one placebo matches the investigational drug, while a second placebo matches the blinded commercial comparator. The placebo approach will influence the blinding approach, may add analytical work, and is often the most difficult aspect of the decision making around comparative agent strategy. For example, an overprinted commercial tablet is a relatively easy, inexpensive manufacture with very little analytical work. The matching placebo, however, requires the manufacture of a matching placebo tablet printed like the comparator which is then overprinted like the blinded comparator. Again, the placebo doesn’t need much analytical qualification, but it is a rather significant manufacturing expense. An over-encapsulated tablet requires significant analytical qualification and stability work, but requires a relatively quick capsule manufacture for both the comparator and the placebo. These simple examples illustrate that with all that must be considered, there needs to be good communication between the business, technical, and the clinical functions to expedite these decisions so they don’t impact study trials or the quality of the comparative agents/placebos. Once the blinding methodology decisions are made, however, the analytical qualification work will begin.
Qualification Criteria – One Company’s Approach
Comparative Agent does not start as an Investigational Drug. Remember…. the comparator is an approved, marketed drug product.It has been QA released and has stability data to support its labeled expiration date and storage conditions. The blinding method, then, must not change the way the comparator performs so that data generated stands up to challenges from the Innovator, challenges from regulatory agencies, and challenges in the clinic (does not break the blind). Qualification of the blinding method must prove this to be true. Pfizer’s CAD group bases the data needed to qualify on the same categories of reprocessing described by the SUPAC documents, but defining them as Negligible, Minor, and Major (Table 1). The purpose for qualification is to assess performance of the blinded comparator relative to the unblinded comparator. There is no need to re-establish acceptability of the commercial product because it’s already marketed and released. But once blinded, it needs to perform similar to that form that is marketed and released.
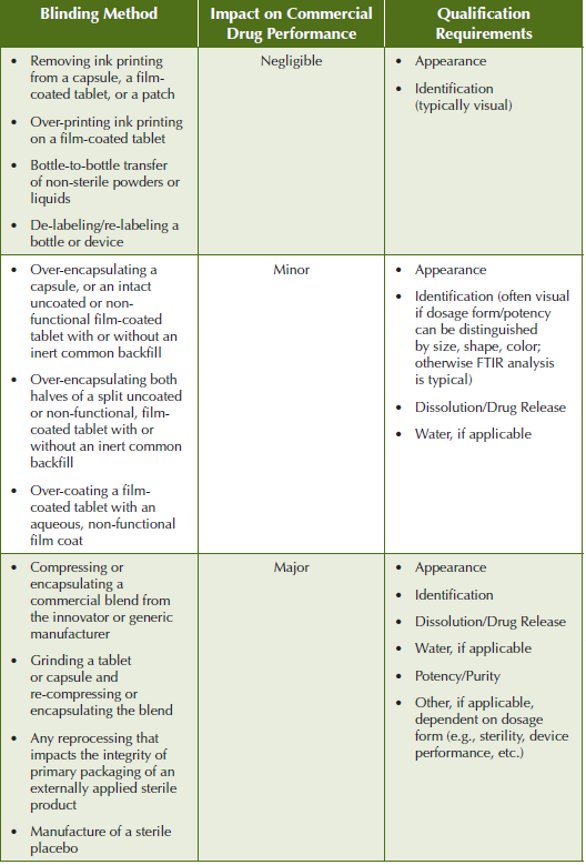
For blinding methods with negligible impact on the performance of the commercial dosage form, the SUPAC guidance states that there is no dissolution documentation needed beyond compendia requirements, and no in vivo bioequivalence is required. Qualification for this category of blinding methods starts with preparation of “check” samples such as deinking or overprinting in the lab to check blinding adequacy/feasibility. The samples are visually inspected for physical/visual defects, discoloration, inability to remove ink, removal of film coating, changes in color of film coating, ghosting, adequate and acceptable appearance of ink over-print. Bottle-to-bottle transfer “check” samples are analyzed for acceptable weight variation or deliverable volumes following transfer of powders and liquids. Repacking checks include visual inspection to assure adequate removal of labels and acceptable appearance of new labels.
The qualification process for blinding methods having minor impact to the commercial dosage form performance starts with preparation of a development lot. If a backfill is used, the establishment of an average weight of the excipient per capsule for the development lot will be used for the manufacturing instructions. These blinding methods with “minor” modifications to the commercial dosage form do require dissolution testing, development of which depends on drug solubility. Multi-media assessment is required for immediate release but not for delayed- or modified-release products. A typical approach for immediate release is to use the three media specified by USP- <711> - 0.1N Hydrochloric acid, pH 4.5 and 6.8 buffers for an initial assessment of the “worst case” potency range, then further develop and validate the final analytical method with the optimal medium. The typical approach for sustained release is to sample in the buffer stage only because the formulation is meant to make it through the acid stomach into the intestinal tract before dissolving. The acceptance criteria for the final dissolution comparison between the blinded and unblinded comparative agent is that the dissolution profiles must be similar. Guidance documents specific to comparative agent dissolution testing are actually specific to bioequivalence, defining the instances when in-vivo data provides assurance of in-vitro drug performance. Three industry documents from the Food and Drug Administration (FDA) and European Medicines Agency (EMEA) give guidance for qualification and dissolution testing [6,7,1], the latter document stating, “Appropriate in vitro dissolution should confirm the adequacy of waiving additional in-vivo bioequivalence testing. Accordingly, dissolution should be investigated at different pH values […] normally pH 1.2, 4.5 and 6.8 unless otherwise justified. Similarity of in vitro dissolution should be demonstrated at all conditions within the applied product series, i.e. between additional strengths and the strength(s) used for bioequivalence testing. […] the applicant could show similar profiles at the same dose (e.g. as a possibility two tablets of 5 mg versus one tablet of 10 mg could be compared)” [1].
Qualification is generally performed on every strength, every source, and every brand (for example an innovator versus a generic) when dissolution profiles are required. Over-encapsulating more than one tablet in a capsule can be difficult to prove bioequivalence, but many in the industry do this routinely, and the EMEA guideline stating, “Medicinal products are pharmaceutically equivalent if they contain the same amount of the same active substance(s) in the same dosage forms that meet the same or comparable standards. Pharmaceutical equivalence does not necessarily imply bioequivalence as differences in the excipients and/or the manufacturing process can lead to faster or slower dissolution and/ or absorption” [1] may also suggest the acceptability of this practice if bioequivalence can be proven.
An additional industry document from the Federation International Pharmaceutique (FIP), meant to be scientific recommendations rather than regulatory guidelines, gives excellent guidance on methodology development and determination of acceptance criteria [8], but the most widely known Comparative Dissolution Criteria is found in the SUPAC documents where “similar” is simply defined as “having a general likeness” [3,4]. One must turn to the bioequivalence documents to find that “general likeness” can be mathematically determined with the calculation of a “similarity factor” called “f2” as follows:
f2 = 50 LOG {[1+1/n Σnt=1 (Rt-Tt)2]-0.5 x 100}
Where, R and T are the percent dissolved at each time point, and an f2 value between 50 and 100 suggests the two dissolution profiles are similar.
It can be established mathematically that when all individual dissolution result values are >80% dissolved at 15 minutes (meet USP Stage 1 where Q=75% at 15 minutes), or the absolute difference of the mean percent dissolved at each time point is <10%, the f2 criteria are met.
The final parameter for qualification of “minor” modifications is to evaluate changes in moisture levels. However, the CAD group has determined over many years that moisture determinations throughout a stability study are only required if the dosage form is susceptible to moisture, which is usually indicated clearly on the package, though the use of a desiccant in the packaging is an indication as well.
Qualification for blinding methods having “major” modifications to the commercial dosage form might start with preparation of a development lot or even performance of a pilot run. Dissolution comparisons are required if they are applicable to the dosage form involved, and are conducted per the discussion above for “minor” category changes, as are moisture level evaluations. In addition, assay/purity and other “function” testing may be needed. Sterility, preservative content and container-closure integrity should be considered for sterile dosage forms, microbiological and particulate analysis should be evaluated for most liquid, ophthalmic, and inhalation dosage forms, and if delivery or other medical devices are used, device performance assessment may be required, for example Emitted Dose and Fine Particle Dose/Distribution (i.e., NGI, Next Generation Impactor, an instrument used for testing spray dispersion) for inhalation devices. These are assessed on a case-by-case basis as they are obviously dependent on dosage form and degree of modification in the blinding process. In regards to devices, in many cases the modification of the commercial product will be a minor de-/re-label, but manufacture of a matching placebo will need extensive product and device performance testing.
Analytical Method Development for Blinding Qualification and Comparative Agent Analysis – A Final Piece of the Puzzle
There are several method development points that should be discussed as they relate to the methods used for qualifying the acceptable blinding of comparative agents. The Pfizer CAD group has recently developed a streamlined process which starts with an information search of the Compendia (including monographs, FDA dissolution database, Pharmacopeial Forum, Pharmeuropa), the commercial Registration Dossier, innovator documents available through Freedom Of Information (FOI), patents, and other literature (journals, package inserts, Merck Index). Then they obtain samples of commercial product and excipients, have package inserts translated if necessary, make prototypes of the blinded product as discussed previously, and then determine the methods needed for qualification, release and stability, including chemical, microbiological, and physical methods. These might be compendia methods which are verified that they work for the blinded product before validating. However, even if available, compendia methods are not required to be used if they are not appropriate for blinded product, but they can still be a good starting point. Often the methods need to be developed. The learning that occurs during method development may influence the validation parameters and acceptance criteria, but both must be documented scientific rationale. Elements of the dissolution development analyses may be applied to the validation requirements if justified. For example, the preparation analyzed during the comparative dissolution run can also be used as one repetition in the reproducibility analysis. However, dissolution, assay, identification, and purity methods all need specificity determined during development before proceeding to validation.
A typical process for development and validation of dissolution, assay, and purity methods is to determine and optimize the end-analysis, often HPLC. A generic Reverse-Phase C18 gradient method with UV detection can be used, with no refinement necessary if the peak shape and separation are acceptable. For immediate-release dissolution the best media is determined first with one source/one strength, and once the appropriate media is selected, the validation and comparative dissolution is performed on all the sources and strengths to be used in the clinical studies. Water methods are only developed when the commercial product has an indication of moisture sensitivity such as a specific label indication, or a primary package that indicates moisture sensitivity such as a foil/foil blister or a bottle that contains desiccant. If required though, the method development again starts with generic volumetric Karl Fischer analysis following USP guidelines. Acceptable stir time, sample volume, etc. are determined prior to validation. The validation has been streamlined to five sample runs: three to determine repeatability, one to determine recovery, and one to determine specificity. The specificity determination is performed at three times the nominal sample concentration to uncover reactions with the iodine/iodide titrant. Coulometric water analysis is rarely needed.
The guideline for identification can be found in the EMEA’s IMP guide which states, “Where an intact solid oral dosage form that is easily identifiable by its colour, shape and marking is encapsulated, identification of the active substance may or may not be necessary, and visual examination may suffice for identification. […] The specifications [for placebo] should at a minimum include a test which enables to clearly differentiate between the respective investigational medicinal product and the placebo.” [5] In other words, when over-encapsulated, the commercial tablet can be thought of as the “active ingredient” for purposes of required identification testing. If one “active ingredient” and its potency can be distinguished from another, and the “active” distinguished from the placebo through visual inspection of appearance, no chemical identification or potency analysis is needed.
Rarely will guidelines for development and validation of other methods be needed. These might include Microbiological methods for Antimicrobial Effectiveness Testing (AET), Sterility, Bioburden, Microbial Limits, or Total Anaerobic Microbial Contamination/Total Yeast and Mold Contamination (TAMC/TYMC), Sub-visible Particulate Matter methods (Light Obscuration or Microscopic), and Device Performance. Many of these have compendia guidelines but are generally developed and validated on a case-by-case basis.
Project Wrap-Up
More specific regulatory guidelines for developing and qualifying blinded comparative agents are finally being published, but the best decisions are based on historical data, scientific judgment, and years of experience in blinding methods for commercial comparators. The Pfizer Comparative Agent Development group, along with their supply, manufacture, and clinical colleagues, has this strong foundation for their comparator strategies. However, they know the industry and regulators change as they also develop additional data and gain experience and scientific knowledge. The pharmaceutical industry is a fast-paced, everchanging, but exciting industry to work in.
References
- European Medicines Agency (EMEA), CPMP/QWP/EWP/1401/98 Rev. 1: Guideline on the Investigation of Bioequivalence, 20 January 2010.
- European Commission, Volume 4 Good Manufacturing Practices Annex 13: Manufacture of Investigational Medicinal Products (July 2003, updated February 2010).
- Food and Drug Administration Center for Drug Evaluation and Research, Guidance for Industry: Immediate Release Solid Oral Dosage Forms, Scale- Up and Post Approval Changes Chemistry, Manufacturing, and Controls, In Vitro Dissolution Testing, and In Vivo Bioequivalence Documentation (SUPAC-IR, November 1995).
- Food and Drug Administration Center for Drug Evaluation and Research, Guidance for Industry: Modified Release Solid Oral Dosage Forms, Scale-Up and Post Approval Changes Chemistry, Manufacturing, and Controls, In Vitro Dissolution Testing, and IN Vivo Bioequivalence Documentation (SUPAC-MR, September 1997).
- European Medicines Agency (EMEA), CHMP/QWP/185401/2004 final: Guideline on the Requirements to the Chemical and Pharmaceutical Quality Documentation Concerning Investigational Medicinal Products in Clinical Trials, Committee for Medicinal Products for Human Use, 31March2006.
- Food and Drug Administration Center for Drug Evaluation and Research, Guidance for Industry: Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System (August 2000).
- European Medicines Agency (EMEA), CHMP/EWP/213035/2007: Concept Paper on BCS-based biowaiver, 24 May 2007. 8
- Federation International Pharmaceutique (FIP), Guidelines for Dissolution Testing of Solid Oral Products (Final Draft, 1995).
Karen Back-Moore has extensive experience in the pharmaceutical industry. She recently was named Technical Learning and Capability – Quality Project Lead in Pfizer Global Supply. Between 2006 through 2010 she was a Principal Scientist in Pfizer Global Research and Development (PGRD) in Groton, Connecticut where she had a variety of roles responsible for the testing of clinical supplies in all phases of study, especially comparative agents and liquid dosage forms, both small and large molecule. Previous to her move to PGRD, Karen held roles of increasing responsibility within Pfizer's Quality Operations organization. Karen has a bachelor degree in Chemistry from Avila University and a master degree in Organizational Management from the University of Phoenix, both in Kansas City, Missouri. She is currently pursuing certification in project management through the Boston University Corporate Education Center.