Support Statement and Disclaimer
All authors are currently employed by Eli Lilly & Co. and contributed equally to this document. None of the authors were supported by external grants.
Introduction
The Pharmaceutical Industry is in the midst of significant change as it looks to balance the cost of developing new medicines while meeting the expectations of patients, healthcare professionals, payers, regulatory agencies and shareholders [1]. The majority of large Pharmaceutical companies originated from bulk chemical manufacturing companies with significant chemical synthesis and production infrastructure. In the 40s, the discovery of penicillin and other innovative medicines revitalized the synthetic chemicals divisions within these companies and institutionalized pharmaceutical Research and Development. The industry expanded in the 60s, benefiting from increasing consumer affordability, high barrier to entry from the cost of R&D and a favorable environment for patent protection. In the last decade however, increasing competition, patent limitations, regulatory reform and the resulting wave of mergers and acquisitions has created the need for increased productivity at reduced cost. Separately, the growth of generic drugs and the expiration of key patents expected to peak between 2010-2014 is estimated to put more than $200 billion in annual sales at risk each year [2, 3]. Challenges in the Pharmaceutical industry are not unfamiliar; less than one in ten molecules tested in man may eventually yield a drug, yet sequencing the human genome has uncovered a vast area of biology, identified numerous potentially “drugable” targets and ushered in the era of Pharmacogenomics. Simultaneously, a number of innovative satellite companies have emerged specializing in reagents and instrumentation for molecular, biochemical and protein analysis, creating unique opportunities for collaborative research. Once again, the Pharmaceutical industry appears invigorated and poised to take advantage of the external environment and support the innovation of successful products. Much like the automobile and aerospace industries before, the archetype of a centralized Pharmaceutical with ownership of all parts ofR&D capability has transitioned to an outsourcing-based model where risk and cost are shared and spread across partnerships.
This shift from a centralized Pharmaceutical R&D to partnered innovation, development and commercialization has also created the need to manage and monitor the clinical trial engagement process of partnered projects. Traditionally, major pharmaceutical companies have pursued the discovery, development, manufacture and commercialization of medicines largely by owning and controlling each component. Tasks previously performed under the umbrella of discipline-based departments that functioned much like their academic counterparts are now organized for maximal efficiency and managed separately by several small companies each with select expertise and service specialty. In this article we focus on an approach to effectively outsource and manage two key areas in the translation of biomarkers into clinical trials; partnering with external laboratories to develop and implement biomarkers and the centralized laboratory concept to manage trial logistics and data collection in support of early phase clinical development decisions. For the purpose of brevity, the scope of this discussion is limited to markers that are not intended to support toxicity or related adverse effect but it is understood that safety markers are included in all clinical studies and do have an essential role in clinical development decisions [3]. The practice of outsourcing pharmacodynamic and pharmacogenetic biomarker testing, its development and implementation in clinical trials discussed here is equally essential to successful transition of a new molecular entity(NME) from early to pivotal stage in the drug development process and can guide critical portfolio decisions based upon probability of technical success (pTS). As stated in a recent perspective, the use of biomarkers in phases I and II can lead to earlier discontinuation of poor performing molecules at a time when fewer resources have been invested [4]. The estimated effect of delays in achieving pTS in Phase II/III of clinical development can add as much as 60% to the capitalized cost at launch, now estimated at $1.8 billion per molecule [3]. Here we outline the essential parts to enable esoteric biomarker assay performance in support of clinical trial protocol objectives, while maintaining technical and regulatory expectation oversight.
The Landscape of Technical Capability and Partnership Opportunities for Phaseappropriate Biomarker Development
A major goal in Translational Medicine is to facilitate the transfer of esoteric markers to support assessment of a pharmacodynamic effect, verification of mechanism of action from pre-clinical models in a clinical setting, or select a patient pool most likely to benefit from the intended treatment. As the role of these biomarkers has elevated from biological curiosity “exploratory” to supportive in clinical development decision, so has the need for assay performance in a regulatory approvable setting. The proper use of statistical methods is another important consideration at this stage as many specialty service providers tend to grow from a research setting and may not be adequately prepared for assay validation in the clinical setting [5]. In most cases, the expectation is to have such analytical measurements performed with the same validation rigor applied to approved clinical tests routinely performed in a Clinical Laboratory Improvement Amendments (CLIA)-certified clinical laboratory. Others feel that the intended use of the data produced by any assay performed on patient samples should determine the level of assessment of variability associated with the analysis “fit for purpose” validation. The foresight of such intended use is often underestimated, leading to inappropriate selection of the analytical platform or an outsourcing partner unable to meet clinical sample testing needs.
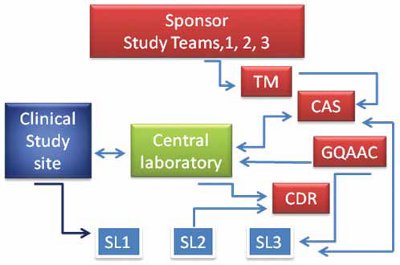
Figure 1- A schematic of the interactive relationships for Pharmacogenomic and clinical trial biomarker support. Sponsor’s study teams interact with Translational Medicine (TM), Clinical Assay Service (CAS), Global Quality Auditing and Compliance (GQAAC) to facilitate Clinical study sites and enable the Central laboratory and multiple Specialty Laboratories (SL) manage sample collection and analysis. Clinical, pharmacogenomic and biomarker data are transferred to Sponsor’s Centralized Data Repository (CDR).
Figure 1 illustrates the core of the interrelationships and accompanying dependencies between the individual specialty laboratories (SL), a centralized Clinical Research Organization (CRO); sometimes referred to as the central laboratory, and the study sponsor. Operation excellence and regulatory oversight are key sponsor-managed activities provided by Clinical Assay Services group (CAS) and Global Quality Auditing and Compliance (GQAAC) groups; individuals with deep clinical operations expertise and knowledge of regulatory expectations for clinical sample testing and data generation. The clinical assay services group would primarily interface between each specialty assay provider and the Clinical Research Organization. The Quality group would assure each specialty laboratory and the central laboratories maintain appropriate regulatory compliance.
Centralized Assay Services Meets Clinical Team Expectations while Handling Test Logistics
The clinical assay services group would play a key role in providing the operational means and technical expertise to deliver test data that is validated, combinable, and accessible while making certain that the data is reportable and reviewable by global regulatory bodies in a timely and costeffective manner. A key role for the associate in this group would be to liaise between all vendors in defining protocol needs for translation to vendor specifications and final data transfer to the sponsor. The range of analysis and activities may include electrocardiograms, imaging, computerized tomography, magnetic resonance imaging, bone mineral density, routine clinical assays, special diagnostic biomarkers, cytology, histopathology, histomorphometry, sample banking and sample storage. The process would begin with partnering with a central laboratory to create kits for collecting protocols’ blood-based diagnostics. Specific assay sample requirements would be gathered from the assaying laboratories that may include type and size of collection tube, processing instructions, storage and shipping requirements, and format of results. Once the kits are created and shipped to clinical study sites, samples would be collected, processed and shipped to the central laboratory. Based on the analysis requirements, samples may then be batch-shipped to specialty laboratories according to a pre-defined shipment frequency. The specialty laboratory would typically transmit results to the central laboratory, and these results would then transmit to the sponsor. The process would be continually reconciled and annotated with a case report form for each subject while on study.
Each step of working with vendors would be standardized to gain efficiency, with regular communication between the clinical assay groups associate, sponsor team and specialty laboratory to ensure the protocol is fulfilled, as well as any unanticipated changes from protocol amendments are documented and managed without enrollment delay. Efficiencies would be gained within a therapeutic area, across multiple protocols for a compound or similar compounds, through consistency over time. The development of “best practices” from collaborations is another key element in optimizing efficiencies even though the broader team members are from multiple, independent companies. As point person for all the vendors, the clinical assay group associate would provide pivotal learning and operational leadership to support the protocol’s vendor-based diagnostic testing.
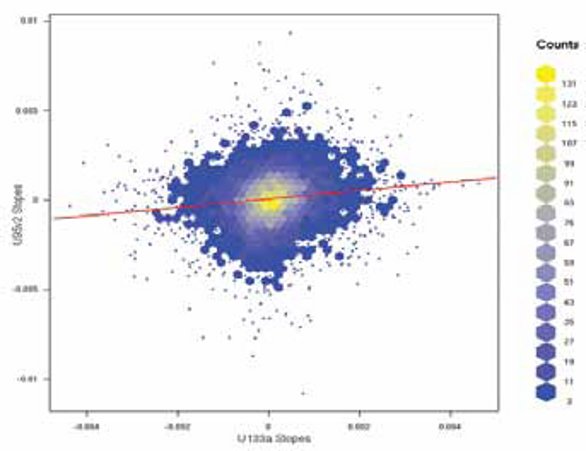
Figure 2- Representation of signal correlated intensities for identical probe sets from two versions of microarray chips used for bridging from an older array to a newer array. The data represented by signal counts shows no systematic error and would be considered acceptable for transition to the array version.
The Evolving Role of Global Quality Auditing and Compliance Management
Global Quality Auditing and Compliance would be an independent quality unit to provide assurance of Sponsor’s compliance with internal quality standards and regulatory expectations. Some of the regulations that guide its function are Good Clinical Practices, Country Specific Government Regulations, Ethics Committees, Good Laboratory Practices, Local Standards (including company policies, company brand attributes), Principles of Medical Research, Ethical Interactions with Health Care Providers, as well as multiple guidance and best practices documents issued by regulatory agencies [6]. The unit would achieve these expectations through continued on- and off-site evaluations and audits of Specialty laboratories’, technical and clinical readiness assessments before study initiation and periodic audits after sample analysis start-up and completion. Assessments and audits would be followed up with documented findings and corrective action and preventative actions (CAPA), as well as documentation and verification of resolved issues allowing progressive seamless integration for clinical analysis of multiple study markers. As the technical complexity of genomic analysis methods and the resulting volume of data from RNA- and DNA-based analytical measurements increases so does the need for quality in managing the fidelity of data transfer and consistency in interpretation. Adoption of well-annotated data standards has the potential to decrease vendor startup time through the creation of a library of reusable data collection and analysis tools. Separately, assay validation experimental design should include built-in reaction and batch controls for assessment of longitudinal performance. The measurement of “precision” with multiplexed analysis such as Multi-analyte immune panels, RNA microarrays and mixed quantitative PCR may require charting periodic measurements performed with a “clinical sample-like” matrix. As illustrated in Figure 2, whenallowing progressive seamless integration for clinical analysis of multiple study markers. As the technical complexity of genomic analysis methods and the resulting volume of data from RNA- and DNA-based analytical measurements increases so does the need for quality in managing the fidelity of data transfer and consistency in interpretation. Adoption of well-annotated data standards has the potential to decrease vendor startup time through the creation of a library of reusable data collection and analysis tools. Separately, assay validation experimental design should include built-in reaction and batch controls for assessment of longitudinal performance. The measurement of “precision” with multiplexed analysis such as Multi-analyte immune panels, RNA microarrays and mixed quantitative PCR may require charting periodic measurements performed with a “clinical sample-like” matrix. As illustrated in Figure 2, when like” matrix, to assess performance especially in instances where an idle biomarker assay is revived multiple times during batched clinical sample analysis from a single study. Significant deviation from a predefined acceptance range can highlight when the assay was not within the acceptable range. When a review of such control charts indicates the process is under control (i.e., is stable, with variation only coming from sources common to the process) then data from the process can be used to predict the future performance of the process. The quality management process as well as Sponsor teams, benefit from such assay performance mapping practices. They help spot deviations and monitor reference/ tolerance ranges.

Figure 3- Illustration of the Cycle threshold value for a control gene charted over a two year period of quantitative RT-PCR analysis performed on accrued batchanalyzed study samples.
Conclusion
Drug development in the era of biomarkers is a complex process that requires a broad array of medical, technological and laboratory expertise. Strategic outsourcing through a centralized laboratory, ideally with global presence, can provide access to additional therapeutic and regional regulatory expertise, extensive clinical drug development experience, and clinical sample handling technologies without adding the associated fixed costs to the Sponsors’ R&D overhead. It meets the need for speed critical for improving pTS in the early phases of development that is especially important in a multinational clinical trial setting. Leveraging these external technical and process-driven resources to produce high-quality data in a timely manner and provide clear protocol-driven decisions is at the heart of a Sponsor’s capability to select and manage partnering relationships. While the model relinquishes everyday decisions and operational control to external partners, it must alleviate this risk through the judicious use of quality audits. Much of the effort in providing the essential oversight remains in the background when such partnerships are successful. On occasions where the reliability of specific parts of the outsourcing process are questioned, pre-study audits and follow-up documentation become an important part of tracking deviations and implementing resolution. People skilled in technologies and communication appear to readily grasp this approach, essential for operating effectively across organizational boundaries.
Integration of Sponsor biomarker strategies with external partners can further improve efficiency as each is able to anticipate needed resources. As more clinical studies are performed globally, especially in countries like India and China, the partnership-based outsourcing approach would release Sponsor resources that would otherwise become necessary to operate regionally. Where local regulations prohibit DNA sampling and/ or export, the central laboratory would be a critical link to local service providers for access to quality guided biomarker and pharmacogenetic analysis and data collection. As the trend in the industry continues to focus on cost-effective drug development, the role of Translational Medicine in partnering with central assay services to deliver biomarkerguided portfolio decisions could become the cornerstone for successful pharmacogenomics integration.
Acknowledgements
The authors acknowledge Lilly colleagues Dr. Stephen Eck and Dr. Michelle Penny for critical review and insightful discussion. We thank multiple external partners for professional engagement while increasing our collective efficiency in delivering much needed medicines in a costconscious environment.
References
- Cockburn IM. The changing structure of the pharmaceutical industry. Health Affairs, 2004, 23:10-22.
- Brody H. Hooked: ethics, the medical profession, and the pharmaceutical industry. Rowman & Littlefield Publishers
- Steven M. Paul, Daniel S. Mytelka, Christopher T. Dunwiddie, Charles C. Persinger, Bernard H. Munos, Stacy R. Lindborg and Aaron L. Schacht. Nature Reviews, Drug Discovery. 2010, 9:203-214.
- Eck SL and Paul SM. Biomarker qualification via public-private partnerships. Clinical Pharmacology and Therapeutics, 2010: 87, 21-23.
- Altshuler JS and Altshuler D. Organizational challenges in clinical genomic research. Nature, 2004:429, 478-481.
- Pocock, SJ. Clinical Trials, a practical approach, John Wiley & Sons, 1993, 187-206.
Dr. Sunil Kadam, Ph.D, Biochemistry & Enzymology, University College, Dublin, Ireland has 25 years of experience in the Pharmaceutical Industry ranging from Discovery, Pharmacogenomics to Translational research. He is currently involved in the development, validation and outsourcing of esoteric biomarkers that support early phase Oncology molecules. He has developed several successful partnerships to facilitate specialty labs and clinical services providers with the integration of analysis and data to support clinical drug development decisions. His educational experience includes Post Doctoral Fellowships in Genetics (University of Calgary, Alberta, Canada) and Biochemical Genetics (Massachusetts Institute of Technology, Cambridge, MA)
Robert Lykins, MS, Chemistry, BS Computer Science, Butler University, Indianapolis, IN, has over 25 years experience in the pharmaceutical industry ranging from laboratory automation projects in Pharmaceutical Product Development to Quality Assurance. His current assignment is with the Global Quality Auditing and Compliance group where he provides auditing support for laboratories, central readers and computer systems.
Catherine Leppert, MT (ASCP) SH, is an Associate Consultant Clinical Diagnostic Services with Eli Lilly and Company and has worked exclusively to support development of the Oncology therapeutic area molecules during the clinical trial phases. Catherine holds a Bachelor of Health Sciences summa cum laude in Clinical Laboratory Science from University of Missouri. Prior to joining Eli Lilly and Company in 1998 she was Laboratory Supervisor at Missouri Cancer Associates in Columbia, Missouri.
Anita Pascarella-Hallett, BS, is the Director of Global Clinical Diagnostic Services at Eli Lilly and Company and supports all therapeutic areas and phases of development with regard to global biomarker operations and central sample storage. Anita holds a Bachelor of Science degree in Biology from Indiana University. Prior to joining Eli Lilly and Company in 1999, she was the Testing Laboratory Manager at Indiana Blood Center in Indianapolis, Indiana.