Trevor Williams*- Pii, A Jabil Company *Corresponding author; Kim Huynh-Ba - Pharmalytik, LLC
Abstract
The current effective version of ICH Q14-Analytical Procedure Development was adopted on 1 November 2023. Development of analytical procedures under the new ICH Q14 guideline poses significant challenges yet also opportunities for the pharmaceutical industry. From analytical target profiles (ATP) to method lifecycle management, the guideline encourages a structured, risk-based approach that integrates Quality by Design (QbD) principles and analytical advancements into method development activities. Addressing these challenges effectively ensures robust, reliable, and compliant analytical procedures essential for drug development, regulatory approval, and global commercialization. This paper explores the critical aspects of method development and validation under the enhanced approach outlined in the Q14 guideline, emphasizing the key principles and expectations while highlighting strategies to navigate these complexities, optimize efficiency, and enhance pharmaceutical product quality and patient safety.
Introduction
With the continuing advances in the pharmaceutical sector, there is increasing demand for robust, reliable, and compliant analytical methods that ensure the safety and efficacy of medicinal products and devices. The desire to better organize and manage analytical method development and the method lifecycle was first evident upon the introduction of the ICH Q8 and Q9 guidelines. Analytical scientists were applying the concepts of these two guidelines to method development, creating their own terminology, and extrapolating the QbD and risk management concepts of the drug development process to analytical methods.1,2 In time, understanding the existing interest, the ICH committee began building a guideline specifically for method development to provide a more structured and systematic approach to analytical method development, incorporating concepts from ICH Q8 and Q9 into the development process of analytical methods. The draft Q14 guideline was initially endorsed in March of 2022 and, after being open to the public for comments and subsequently finalized, was adopted by the regulatory members of the ICH assembly in November 2023.
Since 2023, there have been many challenges associated with adopting the enhanced ICH Q14 framework, particularly in the context of integrating these principles into existing methodologies and pharmaceutical quality systems (PQS). Addressing these challenges ultimately contributes to the development of more effective and reliable pharmaceutical products by minimizing measurement variation and thus helping minimize the total variation in the overall pharmaceutical development process. 1
Analytical Procedure Development
Integrating QbD into analytical procedure development under ICH Q14 requires a shift in mindset and methodology. Under the traditional approach (termed the minimal approach), analytical methods are developed with a focus on meeting immediate performance criteria obtained from established prior knowledge of previously implemented methodologies, standard operating procedures (SOPs) governing such activities, limited experimentation, or a combination of the three. However, QbD emphasizes a deeper understanding of the method’s performance throughout its lifecycle, demanding a proactive, systematic approach to identifying and controlling measurement variability. This requires not only the utilization of prior knowledge but extensive experimentation, risk assessment, and the establishment of robust analytical target profiles (ATPs). While QbD enhances method robustness and flexibility, it necessitates significant investment in time, resources, and expertise, which can be challenging for organizations accustomed to more conventional approaches.
Method Development Strategies
The ICH Q14 guideline advocates for a structured and science-based approach to analytical method development, emphasizing the importance of understanding and controlling variability throughout the method’s lifecycle. The general flow of this procedure lifecycle under the Q14 guideline is depicted in Figure 1.
The ATP is an integral piece of the Q14 framework from which the course of method development is determined. The goal of the ATP is to provide an outline for what specifically is being measured, the criteria that a particular measurement must be able to meet to ensure quality of the data, and is derived from specific critical quality attributes (CQA) summarized in the quality target product profile (QTPP). Prior knowledge, established criteria via regulatory guidelines, and product/process understanding contribute to the formation of an ATP.3 It is possible for multiple technologies and approaches to satisfy a given ATP. Thus, part of the challenge with utilizing an ATP for method selection is the amount of initial experimentation that may be necessary to scout and evaluate potential candidate methodologies if sufficient prior knowledge is lacking.1,3 However, establishing an ATP can become a major advantage of the enhanced approach to analytical procedure development, giving flexibility to technology selection.

Once the proposed technology has been selected for method development, QbD principles such as Design of Experiments (DoE) guide the identification of Critical Method Parameters (CMPs) and their Proven Acceptable Ranges (PAR) or Method Operable Design Regions (MODR) with their impact on method performance, allowing for the setting of Established Conditions (ECs). QbD aims to establish a design space detailing and guaranteeing method performance.4 The amount of experimentation and data management can be extensive, requiring laboratory resources; however, it will be a well-built repository for development results, allowing for ongoing knowledge management.3,4
In addition to the QbD approach, risk assessment is critical throughout the method development process to determine method parameters that would impact the method performance, as well as method changes. Tools such as Ishikawa diagrams and Failure Mode and Effects Analysis (FMEA) help assess and categorize risk, driving decision making and ultimately leading to the development of analytical control strategies.5 Risk assessment is performed at the outset of the development process and then repeated during development based on the data available. Risk assessment can be either an informal or formal exercise.3 Often, it is best to utilize such tools as a formal exercise to allow for knowledge capture and management, setting precedents for possible future use in a good manufacturing practices (GMP) environment.
By adopting these strategies, in part or in full, organizations can develop analytical methods that are robust, compliant with regulatory expectations, and capable of delivering reliable results throughout their lifecycle.
Analytical Control Strategies
An analytical control strategy is essential for ensuring method reliability throughout its lifecycle. Under ICH Q14, this strategy involves identifying potential sources of variability, whether that be system, user, or environment related, and implementing controls to mitigate their impact using system suitability and sample suitability tests and other established conditions.
Under the enhanced Q14 approach, understanding what constitutes an established condition (EC) can be challenging for laboratories initially implementing the Q14 guideline.6 The concept of ECs was first described in ICH Q12. In regard to analytical procedures, they are described to be performance characteristics and criteria, procedure principles, e.g., the specific technology used, system suitability, and sample suitability criteria, and set points or ranges required for procedure parameters.3,7 Included with ECs are risk-based categorizations of their impact on method performance along with the rationale for their set points and ranges, requiring the developers to elaborate on the logic behind decisions made. Rationale listed for ECs may be succinct but often require extensive prior knowledge and experimental data for support unless they are dictated by regulatory guidance.3,7
Another aspect of control strategy is the establishment of continuous monitoring and feedback loops, which ensure ongoing method performance. Continuous monitoring reduces the likelihood of method failure and supports consistent, high-quality results in routine analysis. The benefit of continuous monitoring is the ability to quickly discover out-of-trend method outputs (OOTs) in overall method performance, thus lowering the likelihood of method-related investigations. Such changes in performance can then be investigated for potential impact on the results and facilitate root cause analysis for any observed Out-of-Specification results (OOS), potentially decreasing the probability of batch release failures and product recalls. The challenge with continuous monitoring is proper data management to allow for the trending of the appropriate method outputs or system suitability test (SST) results. Many LIMS software programs are capable of inputting and trending SST results as part of the overall control strategy. Such data can also be valuable to improve the robustness of the method or accommodate changes in analytical technologies.
Analytical Procedure Lifecycle Management
Analytical lifecycle management (ALCM), a core principle built into the ICH Q14, is extrapolated from the ICH Q12 guideline for pharmaceutical product lifecycle management.7 While the ICH Q12 is mainly focused on direct changes to commercial drug products via formulation and/ or process changes, in Q14, lifecycle management pertains specifically to establishing critical analytical criteria through method development and managing changes in the analytical procedure over the lifecycle of said product. Similar to Q12, these changes are typically post-approval changes occurring after the drug product has already received approval by the relevant regulatory authorities and is in commercial manufacturing. Any changes that are to be made to the procedure must be assessed for potential impact on the reliability of the results and, thus, the product quality and safety. In some cases, an entirely new analytical procedure may be required.
ECs play an important role in ALCM. Because reported ECs are considered legally binding, any changes to them must be justified and evaluated based on their risk categorizations, the level of comprehension of the relationship between method parameters and overall method performance, method intricacy, and current control strategies.3,7 If a particular EC has a high risk of impacting the overall method performance, it is advised to detail the planned change and the studies required to evaluate the potential impact through the use of a post-approval change management protocol (PACMP). PACMPs can be filed with the original submission or as an addendum to the drug product submission.
Understanding the level of risk with each EC requires comprehensive in-depth knowledge of method performance. The benefit of the lifecycle approach of Q14 is regulatory flexibility. ECs that are either PARs or MODRs of method parameters, or if the EC is categorized as low risk/impact, any changes made within the stated ranges or other low-impact changes only require notification to regulatory authorities instead of requiring prior review and approval by said authorities. Under the traditional minimal approach with an absence of evaluating and categorizing ECs based on risk, any changes to relevant method parameters automatically require prior approval from authorities, with supporting evidence in the form of experimental studies and revalidation.
Supporting the lifecycle approach is the continuous monitoring of analytical methods, ensuring they remain fit for purpose over time. As stated previously, this continuous oversight can be resource-intensive and may require the development of new internal processes and documentation systems to ensure compliance. The nature of method lifecycle management involves periodic reviews, assessing the need for improvements, and implementing changes based on scientific rationale and risk assessment. The challenge lies in balancing the need for rigorous lifecycle management with the operational realities of method development and routine analysis. Table 1 shows the typical advantages and disadvantages of ICH Q14 implementation.
Analytical Method Validation
Analytical method validation under the enhanced approach of ICH Q14 emphasizes the lifecycle approach, where validation is not a one-time event but an ongoing process. Methods are validated based on their ATPs, ensuring they meet predefined performance criteria under various conditions. Continuous verification and potential revalidation are required as part of lifecycle management, particularly when there are changes in raw materials, equipment, or process conditions. This approach ensures that methods remain fit for purpose over time, maintaining regulatory compliance and product quality.
Under the Q14 enhanced approach, it may not be possible to fully validate the design space. However, it is expected that sufficient development data exists to justify the method-design space with supporting validation data for the expected routine set-points.3

Regulatory Expectations and Compliance
The introduction of ICH Q14 brings heightened regulatory expectations, requiring a more detailed and systematic approach to method development and validation. At the time of this paper, the ICH offers two overall approaches to method development: the traditional minimal approach and the more recent enhanced approach. While the minimal approach is required, utilizing elements of the enhanced approach is highly encouraged.3 Over time, we can expect to see more favor for the enhanced approach, with regulatory authorities expecting a thorough understanding of the method’s performance across a range of conditions, supported by comprehensive data and risk assessments. This can lead to increased scrutiny during regulatory submissions, as agencies may demand more extensive documentation and justification for method choices. We must remember that even though the minimal approach is required, regulatory authorities can request elements of the enhanced approach at any time if given the circumstances, e.g., a high-risk API requiring the tightest of controls. Ensuring compliance with these new requirements requires organizations to invest in training, process optimization, and potentially new technologies to generate the necessary data and maintain high standards of regulatory readiness. Navigating these expectations while maintaining efficiency and innovation poses a significant challenge for the pharmaceutical industry.
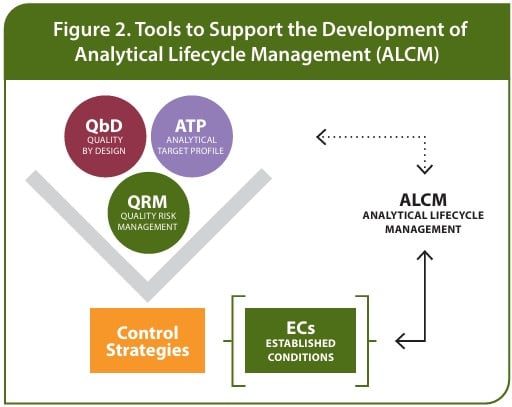
Figure 2 shows the impact of QbD and ATP principles on establishing control strategies for the analytical method performance. This database and knowledge gained from method development will contribute to the established conditions to monitor method changes and trigger additional validation or qualifications to assure the intended use of the method and ensure the product is safe and effective.
Adapting Existing Methods to the ICH Q14 Framework
Adapting existing analytical methods to align with the ICH Q14 framework presents its own set of challenges. Many established methods were developed under different guidelines and may lack the comprehensive documentation and understanding required by QbD and lifecycle management principles. Retrospectively applying these concepts to legacy methods can be complex and resource-intensive, as it may involve revalidation, additional experimentation, and the establishment of new control strategies. Companies must balance the need to update these methods with the potential disruption to routine operations and any additional costs involved. The challenge is to achieve compliance with ICH Q14 while minimizing the impact on existing workflows and ensuring the continuity of analytical operations.
For a developing pharmaceutical company, the key to incorporating ICH Q14 into its culture is to utilize the tools that the company already possesses to implement the needed changes in a systematic way. Change control, document control, LIMS, and the various statistical tools available will help with the indoctrination of the enhanced approach outlined in the ICH Q14 guideline. The ICH gives the option to incorporate elements of the enhanced approach, giving companies the flexibility needed to adapt their processes and procedures according to their current capabilities and timelines.
As ATPs are living documents centered on the CQAs of a drug product, it is possible to retroactively incorporate them into the drug product profile.2 Since ATPs signal the start of the method development process, they are the keystone to the enhanced approach toward Q14 implementation. Another benefit is to build the body of data for continuous monitoring of method performance by trending system suitability. Other benefits can be the streamlining of the review and approval process of analytical method changes from the agency.
Conclusion
The systematic and detailed development of methods utilizing QbD and risk management principles calls for conscious proactive steps to organize and manage vast amounts of data with the target of minimizing measurement variability over the lifetime of a drug product. Managing changes to analytical procedures is also crucial under the ICH Q14 framework, ensuring insights into product changes that require analytical methods remain robust and reliable over the lifetime of the drug product. Implementing these strategies not only improves the quality of the analytical methods but also enhances efficiency in sample analysis and investigation. However, applying these principles globally continues to present challenges, particularly in aligning diverse regulatory expectations and practices. Addressing these challenges will be key to maximizing the benefits of ICH Q14 and advancing the quality and consistency of pharmaceutical products worldwide. We can expect to see a bigger push for the enhanced approach as the industry progresses towards real-time release testing and process analytical technology (PAT).
Acknowledgments
The authors thank the following colleagues for their time and effort in providing outstanding technical peer reviews: Margaret Maziarz, Principal Scientist, Waters Corporation; Cathy Sioma, Ph.D.. Director of Quality Control, Pii, A Jabil Company; Qinggang Wang, Ph.D., Scientific Director, Bristol Myers Squibb; and Travis Webb, Chief Scientific Officer, Pii, A Jabil Company.
References
1. F. G., & Kord, A. S. (2010). Development of Quality by Design Analytical Methods. Journal of Pharmaceutical Sciences, 100(3), 797-812. doi:10.1002/jjps 2325.
2. Vogt Patrick Jackson, et al, Using the Analytical Target Profi le to Drive the Analytical Method Lifecycle, Analytical Chemistry 2019 91 (4), 2577-258, DOI: 10.1021/acs.analchem.8b04596 ICH Q14 Guideline: International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH). (2023).
3. ICH Q14: Analytical Procedure Development. Retrieved from the ICH Official Website. ICH Q8(R2) Guideline: International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). (2009).
4.ICH Q8(R2): Pharmaceutical Development. Retrieved from the ICH Official Website
5. ICH Q9 Guideline: International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). (2023). ICH Q14: Analytical Procedure Development. Retrieved from the ICH Official Website.
6. American Association of Pharmaceutical Scientists (AAPS) Virtual Round Table Discussion, “Challenges and Opportunities in the ICH Q14 Implementation,” hosted by the AAPS CMC Community, 26 September 2024.
7. ICH Q12 Guideline: International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). (2019). ICH Q12: Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management: Retrieved from ICH Official Website.
Trevor Williams is a Senior Analytical R&D Chemist within the Method Validation group at Pharmaceutics International Inc. He has over 10 years of experience working in the R&D and QC laboratories. Trevor obtained a Master of Science in Analytical Chemistry from Illinois Institute of Technology (IIT). Trevor is a long-standing member of the American Chemical Society (ACS) and can be contacted at [email protected].
Kim Huynh-Ba is the Managing Director of Pharmalytik Consulting, LLC. Kim is an Adjunct Professor at Temple University-School of Pharmacy and Illinois Institute of Technology (IIT), teaching Quality Audit, ICH quality guidelines, and Good Manufacturing Practices. She is the editor of multiple book volumes, including the “Handbook of Stability Testing in Pharmaceutical Development: Regulations, Methodologies, and Best Practices” (2008), “Pharmaceutical Stability Testing to Support Global Markets” (2010), and “Analytical Testing for the Pharmaceutical GMP laboratory” (2022). Kim is a member of the USP Council of Experts (2015-2025), where she chairs the Small Molecules 4 Expert Committee. She was named an AAPS Fellow in 2020. She can be contacted at [email protected].