By: Priyanka Suresh Ghugare, Department of Quality Assurance, NIMS University, Dr BS Tomar City; Sandeep Kumar, Department of Pharmaceutics, NIMS University, Dr BS Tomar City
Abstract
The contaminants included in Active Pharmaceutical Ingredients (APIs) are attracting increasing attention. As required by numerous regulatory bodies, the purity profile and the impurity profile are now both crucial needs. Any inorganic or organic material, or leftover solvents that are not a part of the therapeutic components or ingredients, that can occur during synthesis or from undesirable compounds that remain with APIs are referred to as impurities in the pharmaceutical industry. Numerous chromatographic and spectroscopic techniques are used to identify contaminants, either alone or in conjunction with other techniques. There are several methods for identifying and describing impurities, such as Atomic Absorption Spectroscopy (AAS), High-Performance Liquid Chromatography (HPLC), High-Performance Thin Layer Chromatography (HPTLC), and Thin Layer Chromatography (TLC). Impurity profiling has made considerable use of conventional liquid chromatography, especially HPLC; its many uses are a result of its sensitivity, cost-effective separation capabilities, and broad range of detectors and stationary phases. One of the most popular methods for identifying leftover solvents is high-performance liquid chromatography. Impurity profiling has been revolutionized by the development of new techniques that allow for both the isolation and structural identification of impurities.
Introduction
The control of pharmaceutical impurities is a fundamental objective in the development of drugs, focusing on the comprehension of the chemical structures of unidentified impurities. This understanding is essential for evaluating toxicological consequences and grasping the mechanisms of formation. API-related impurities, such as degradation and interaction, can affect drug product quality, safety, and efficacy. Classifying sources of impurities is essential for analytical method development and acceptance criteria (Figure 1).1
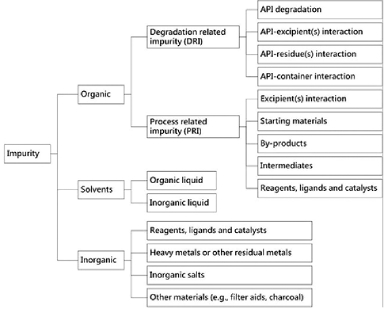
Figure 1. Classification of impurities3
Impurities in drugs must be carefully monitored to ensure quality. Understanding impurities and identifying potential sources is crucial. Selective analytical methods are needed, and profiling impurities can provide comparative data. Isolating impurities when hyphenated methods fail or using authentic materials allows toxicological studies and routine drug product monitoring.2-3
Terminology
This discussion includes various terms used to describe materials affecting API purity, and all considered impurities. It provides an overview of their advantages and limitations.
Identification and qualification thresholds for impurities
The International Conference on Harmonization tackles issues concerning impurities in the following manner:4
- Stability testing for new drug substances and products (Q1A)
- Impurities present in drug substances (Q3A)
- Impurities found in drug products (Q3B)
- Impurities: residual solvents (Q3C)
Specifications: testing procedures and acceptance criteria for new drug substances and new drug products; chemical substances
The guidelines set forth by the International Conference on Harmonization (ICH) have greatly enhanced the clarity regarding the definition of impurities in novel drug substances. To ensure high purity, a scheme for profiling drug impurity is proposed, and analytical methods are employed for quantification.5
The U.S. Food and Drug Administration (FDA) regulates product safety, characteristics, and purity, which can be affected by impurities. These unwanted chemicals can alter the efficacy and toxicity of pharmaceutical compounds. Quality control checks are crucial for maintaining product purity, and various pharmacopoeias set strict limits on purity and impurity. Modern separation methods simplify the separation and characterization of impurities.6
The FDA has endorsed a unified terminology for monitoring impurities in active pharmaceutical ingredients, developed by the ICH. This guideline was framed with collaboration from the EU, Japan, and the US, ensures consistent data requirements for regulatory agencies. It aids sponsors of New Drug Applications and FDA reviewers in interpreting and implementing regulations, addressing ethical, economic, and competitive reasons.7
Impurities Designation
Typical Words for Impurities
The following terms are utilized by various regulatory agencies and the ICH to characterize impurities:
- Intermediate
- Penultimate intermediate
- By-products
- Transformation products
- Interaction products
- Related products
- Degradation products
Intermediate compounds arise during the synthesis process, whereas penultimate intermediates represent the final stage in the sequence. By-products refer to compounds generated during reactions, which may occur due to overreaction or incomplete reactions. Transformation products pertain to both theorized and non-theorized products, while interaction products can be formed either intentionally or unintentionally. Related products are chemically analogous to drug substances and may exhibit biological activity. Degradation products result from external influences.8
Sources of Impurity
The study presents a comprehensive overview of all classes of impurities, highlighting the significant analytical challenges faced in drug discovery compound characterization, quantization, and detection (Figure-2).
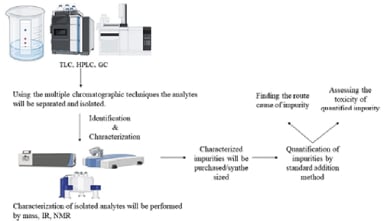
Figure 2. Schematic representation of stages involved in impurity profiling9
Classification of Impurity
The United States Pharmacopoeia (USP) classifies impurities into three distinct sections:
- Impurities in Official Articles
- Ordinary Impurities
- Organic Volatile Impurities
According to the ICH, impurities in drug substances that are produced through chemical synthesis can be generally categorized into the following three groups:
- Organic Impurities (both process-related and drug-related)
- Inorganic Impurities (including reagents, ligands, and catalysts)
- Residual Solvents (which are volatile solvents)10
Organic impurities
Impurities present in the manufacturing and storage of drugs encompass starting materials, intermediate impurities, by-products, and degradation products. Proper care during multistep synthesis is crucial to avoid residual unreacted starting materials. By-products are rare in synthetic organic chemistry, and degradation products can form due to improper formulation storage during bulk drug manufacturing.11
Other Types:
- Impurities caused by synthesis: Synthesis processes generate new chemical entities from raw materials, solvents, intermediates, and byproducts. Impurities can lead to unwanted materials in the final product, requiring careful attention at every step.
- Formulation Related Impurities: Drug substances undergo various conditions, including hydrolysis, which can lead to degradation or reactions, with water in formulations contributing to impurity and hydrolysis and catalysis.12
Inorganic impurities
Inorganic impurities are also derived from the manufacturing processes utilized in bulk drug formulation. These impurities are typically recognized and identified.
- Reagents, Ligands, and Catalysts: The occurrence of these impurities is rare. However, if the manufacturing procedures are not adhered to properly, it may lead to complications.
- Heavy Metals: Water is commonly employed in various manufacturing processes, serving as a primary source of heavy metals such as Arsenic, Cadmium, Chromium, Sodium, Magnesium, Manganese, etc., particularly where acidification or acid hydrolysis occurs. The use of demineralized water and glass-lined reactors can effectively prevent heavy metal impurities.
- Other Materials (Filter Aids, Charcoal): Filtering aids, including centrifuge bags, are routinely utilized in bulk drug manufacturing facilities, and in many instances, activated carbon is also employed, which can contribute to impurity.
Consequently, to prevent contamination, it is crucial to regularly monitor fibers and black particles in bulk drugs.13
Residual solvents
Residual solvents refer to organic or inorganic liquids utilized in the manufacturing process. Completely eliminating these solvents through the work-up process is quite challenging. Certain solvents that are recognized for their toxic effects should be avoided when producing bulk drugs.
Formulation-related impurities (impurities in drug products)
Impurities in drug products can result from inert ingredients and various conditions during formulation, including hydrolysis and catalysis. Water used in formulations can contribute impurities and create a ripe situation for degradation.14
Analytical method development
The development of new drugs necessitates the generation of significant and dependable analytical data throughout various phases of the process.
- Selection of sample sets for the development of analytical methods
- Evaluation of chromatographic conditions and phases, typically employing the linear solvent-strength model of gradient elution
- Method optimization to refine parameters associated with ruggedness and robustness. Impurities can be predominantly identified through the following methods:
- Reference standard method
- Spectroscopic method
- Separation method
- Isolation method
- Characterization method15
Reference Standard Method
The primary objective is to elucidate the life cycle, qualification, and governance of reference standards utilized in drug development and control, which act as benchmarks for assessing drug safety and evaluating both process and product performance.16
Spectroscopic Techniques
The UV, IR, MS, NMR, and Raman spectroscopic techniques are commonly employed for the characterization of impurities.
Separation Techniques
Capillary electrophoresis (CE), Chiral Separations, Gas Chromatography (GC), Supercritical Fluid Chromatography (SFC), TLC, HPTLC, and HPLC are frequently utilized for the separation of impurities and degradation products.
Isolation Techniques
The isolation of impurities is often essential; however, instrumental methods frequently circumvent this step. Both chromatographic and non-chromatographic techniques are employed for this purpose. Chromatographic reactors are analytical-scale columns designed for flowthrough and separation. HPLC was utilized to study the hydrolysis kinetics of the Aprepitant prodrug, fosaprepitant dimeglumine.17
- Solid-phase extraction methods
- Liquid-liquid extraction methods
- Accelerated solvent extraction methods
- Supercritical fluid extraction
- Column chromatography
- Flash chromatography
- TLC
- GC
- HPLC
- HPTLC
- Capillary electrophoresis (CE)
- Supercritical fluid chromatography (SFC)
Analytical Procedures
Method Development: Method development involves selecting columns, mobile phase, detectors, and quantization methods. Factors to consider include accuracy, reliability, cost, time, energy, automation, and analyte selectivity. Newer instrumentation techniques can offer improved methods with enhanced accuracy, precision, and better returns on investments.18
Figure 3. Analytical method validation for quality assurance19
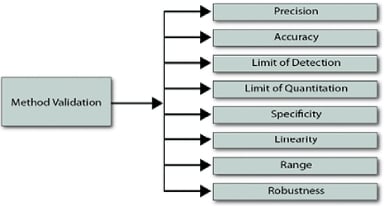
Figure 4. Method of validation20
Validation of Analytical Methods
The validation process confirms a method’s accuracy, precision, sensitivity, and ruggedness for an intended analytical application through laboratory studies and procedures. It provides documented evidence of the system’s systematic, precise, and reliable performance, as per ICH (Figure 3, 4).19
Limits For Impurities
ICH guidelines suggest identifying impurities below 0.1% level isn’t necessary unless potent or toxic. Maximum daily dose qualification thresholds are <2g/day 0.15% or 1 mg/day intake.21
Applications
Pharmaceutical compounds, produced synthetically, naturally, or recombinantly, have numerous applications in drug design, quality, stability, and safety, including alkaloids, amines, amino acids, and more.22
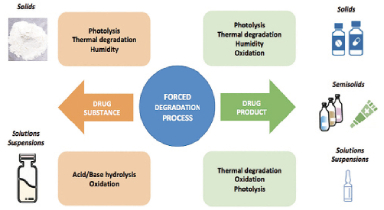
Figure 5. Forced degradation process23
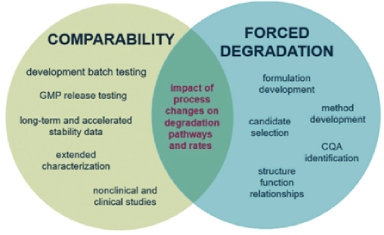
Figure 6. Impact of process changes on degradation pathways and rates24
Forced Degradation Study
Forced degradation studies analyze drug sample stability in pharmaceutical industries, affecting product safety and efficacy. Stability information aids in formulation, packaging, storage conditions, and shelf life. Stability studies are mandatory for registration dossiers. However, there’s a lack of clarity in scientific literature and regulatory guidance on appropriate stress experiments and justifications for declaring a drug stable.23
Forced degradation is a process where drug compounds are subjected to extreme conditions to determine breakdown levels and kinetics. Companies use stress testing practices to validate analytical methods, challenging the specificity of stability-indicating and impurity-monitoring methods. This is part of the development strategy (Figure 5).24
Forced degradation studies examine the specificity of stability in new drug substances and products, providing insight into degradation pathways and products, and elucidating their structure, aiding in formulation and packaging development (Figure-6).
Need for forced degradation of drugs
Research on the forced degradation of pharmaceutical compounds holds significant importance in several areas.
- To establish techniques for assessing stability.
- To identify the pathways of degradation.
- For evaluating the intrinsic stability of drugs within dosage forms.
- To investigate the chemical characteristics of molecules.
- For the creation of stable formulations.
- To ascertain the structure of decomposition products.
- To address issues concerning stability.
- To produce a degradation profile in accordance with ICH guidelines.25
Time to conduct forced degradation
Studies on forced degradation are essential for the development of new pharmaceuticals. The FDA guidelines suggest that stress testing should take place during phase III of the regulatory submission process. These stress studies ought to be carried out in different pH solutions, as well as under conditions of oxygen exposure, light, and increased temperatures. Early-stage stress testing is recommended to identify degradation products, elucidate structures, and optimize the conditions of stress. This helps in manufacturing improvements and stability-indicating analytical procedures selection.26
Limits for degradation
Pharmaceutical scientists debate the appropriate amount of degradation for drug substances. Typically, 5%-20% degradation is acceptable for chromatographic assay validation. However, some believe 10% degradation is optimal for small molecules with 90% stability limits. No limits have been established for physiochemical changes, loss of activity, or degradation during shelf life. Stress testing can be terminated if no degradation is observed, but over-stressing may result in secondary degradation products. Protocols for product-related degradation may differ between drug substances and products.27-28
Selection of drug concentration
The regulatory guidance does not specify the concentration of a drug for degradation studies. It is recommended to start at 1 mg/mL, as minor decomposition products can be detected. Degradation studies should also consider the drug’s expected presence in final formulations.29
Degradation Conditions
Hydrolytic conditions
Hydrolysis is a prevalent degradation process that entails the breakdown of a compound by water in either acidic or basic environments. This method is employed in acid or base stress testing to produce primary degradants. The specific type and concentration of acid or base utilized are contingent upon the stability of the drug substance. Additionally, co-solvents may be incorporated when compounds exhibit low solubility in water. Stress testing should start at room temperature and last no more than 7 days.30
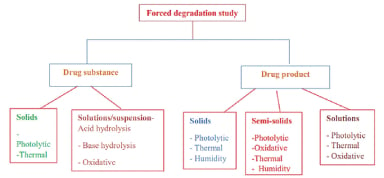
Figure 7. Forced degradation study34
Oxidation conditions
Hydrogen peroxide is frequently employed in forced degradation studies of pharmaceutical compounds; however, alternative oxidizing agents such as metal ions, oxygen, and radical initiators may also be utilized. This process entails an electron transfer mechanism that generates reactive anions and cations. Compounds such as amines, sulfides, and phenols are prone to oxidation, whereas functional groups containing labile hydrogen are also vulnerable.31
Photolytic conditions
Photo stability testing evaluates drug substances’ ability to withstand light exposure without unacceptable changes. ICH guidelines recommend exposure to 1.2 million lx h and 200 W h/m2 light, with the most common wavelength being 300-800 nm, and a maximum illumination of 6 million lx h.32
Thermal conditions
The research suggests evaluating solid-state drug substances and products in both dry and wet heat environments, whereas liquid drug products ought to be subjected to dry heat only. The Arrhenius equation is employed for this analysis.
k=Ae−Ea/RT
where, k represents the specific reaction rate, A denotes the frequency factor, Ea signifies the energy of activation, R is the gas constant (1.987 cal/deg mole), and T indicates the absolute temperature. The thermal degradation study is conducted at temperatures ranging from 40 to 80 degree C.33
The stability of a pharmaceutical ingredient is influenced by numerous factors, primarily including the following:
- Temperature: Elevated temperatures accelerate oxidation, reduction, and hydrolysis reactions, resulting in drug degradation.
- pH: Both acidic and alkaline pH levels affect the decomposition rate of most drugs. The degree of ionization can lead to increased drug degradation.
- Moisture: Water facilitates the catalysis of the degradation process.
- Light: Drug stability is impacted by energy or thermal heat from light, which can cause oxidation of the ingredients.
- Drug Incompatibility: Certain components present in a finished pharmaceutical product may react with each other or with the container’s cover.35
Advantages of HPLC
- HPLC is recognized as the most precise method for separating analytes, impurities, and degradation products.
- The limit of detection for analytes using HPLC is significantly lower compared to other methods, indicating its superior sensitivity when compared to alternative techniques employed in the development of SIAM.
- HPLC requires less time for the separation and quantification of analytes and degradation products.
- It provides enhanced resolution between analytes and impurities, utilizing a wide range of stationary phases.36
- HPLC exhibits greater sensitivity for detecting small quantities of analytes and degradants, as various detectors can be employed to measure the response.
- A notable advantage of HPLC is its reusable columns, which can be utilized multiple times for analysis, rendering it a cost-effective technique.
- The technique is fully automated, allowing for quantification to be performed using this method.
- HPLC has seen extensive application and has become popular in stability studies due to its high-resolution capabilities, sensitivity, and specificity. This technique can also analyze non-volatile, thermally unstable compounds. Consequently, many SIAM have been effectively established using HPLC.37
High performance liquid chromatography
HPLC is a technique in which a solution of a substance is introduced into a column containing a stationary phase, while a mobile phase is circulated through it. This process separates substances according to their migration rates and partitioning characteristics, typically utilizing two main types.38
Normal-phase HPLC
Polar drugs are eluted at a later stage compared to non-polar drugs, where the stationary phase consists of a hydrophilic, water-soluble substance such as silica, while the mobile phase is characterized by being non-polar and fat-soluble.39
Reverse-phase HPLC
RP-HPLC denotes a condition in which the stationary phase exhibits lower polarity compared to the mobile phase, which is usually Octadecylsilane. Alternatives to this include C8 and shorter alkyl chains, as well as cyclohexyl and phenyl groups.40
Need for HPLC method development
The development and application of HPLC methods are essential to drug development in order to satisfy business, regulatory, and scientific requirements in the pharmaceutical sector. It assists in determining possible harmful indications and estimating therapeutic indications. Drug discovery, manufacturing, and quality control all depend on the development and validation of HPLC methods. For safety and quality, impurity identification and measurement are essential.41
Method development
A phase in the development of HPLC methods includes the following steps:
- Comprehending the physicochemical characteristics of the drug molecule.
- Choosing the appropriate chromatographic conditions.
- Formulating the analytical approach.
- Preparing the samples.
- Optimizing the method.
- Validating the method.42
Comprehending the physicochemical characteristics of a drug molecule:
A drug molecule’s physicochemical properties, which include solubility, polarity, pKa, and pH, are crucial in the creation of a technique. Polarity plays a crucial role in determining the solvent and mobile phase composition, while pKa is the negative logarithm to base 10 of the hydrogen ion concentration. The solubility and compatibility of the analyte with the diluents both influence the choice of mobile phase or diluents.
pH = - log10[H3O+].
Proper pH selection for ionizable analytes often results in symmetrical and sharp peaks in HPLC, crucial for quantitative analysis, achieving low detection limits and reproducible retention times.43
Selection of chromatographic conditions:
The selection of a stationary phase or column is crucial for method development since it ensures a high-performance and stable column. A C8 or C18 column made of pure silica is recommended for all samples. The physical characteristics of silica-based packing make it the preferred choice for most HPLC columns.44
Development of approach for analysis:
The RP-HPLC analytical technique requires the selection of chromatographic parameters, including mobile phase, column type, flow rate, and pH, which are determined through trials and system suitability criteria. These criteria encompass retention time, theoretical plates, tailing factor, resolution between peaks, and relative standard deviation (R.S.D.). Typically, the detection wavelength is set at an isobestic point for concurrent estimation. The linearity of the drug is assessed to identify the concentration range that adheres to a linear pattern. Additionally, laboratory mixture analysis is performed.45
Sample preparation:
Sample preparation is crucial in HPLC analysis to create a homogeneous, reproducible solution for column injection. It ensures a solvent dissolve in the mobile phase without affecting retention or resolution, starting from collection to column injection.
Method optimization
The method’s weaknesses are identified and optimized through experimental design, ensuring performance under various conditions, instrument setups, and samples.
Method Validation
Validation confirms method performance, providing objective evidence that specific requirements are fulfilled, particularly at low concentrations, demonstrating method capability.46
Types of Analytical Process validation47
The validation of analytical procedures is carried out by four types:
- Identification assessments;
- Quantitative analyses for the content of impurities;
- Limit assessments for impurity control;
- Quantitative evaluations of the active ingredient in samples of the drug substance or drug product, or other chosen component(s) within the drug product.
Components of method validation48
The following are typical analytical performance characteristics which may be tested during methods validation:
- Accuracy
- Precision
- Linearity
- Detection limit
- Quantitation limit
- Specificity
- Range
- Robustness
Accuracy
The difference between the mean and the true value is reflected in accuracy, which is the degree to which a measured value agrees with the true or accepted value. This is demonstrated by applying the procedure to samples with known analyte concentrations, which are subsequently examined in relation to blank and standard solutions.49
Impurity profiling has made considerable use of HPLC; its many uses are a result of its sensitivity, cost-effective separation capabilities, and broad range of detectors and stationary phases.
Precision
The consistency of measurements made from multiple samplings of a uniform sample under specific conditions is measured by precision. It includes intermediate precision as well as repeatability. While intermediate precision refers to the variance seen in a laboratory context, which may involve multiple days, instruments, and analysts, repeatability refers to the variation seen by a single analyst using a single instrument. Precision and accuracy are not interchangeable.50
%RSD=(Standard devitation×100)/mean
Linearity
The ability of an analytical process to produce a response that is exactly proportionate to the concentration (quantity) of the analyte in the sample is known as linearity. The test results are either directly proportional or can be converted using a precise mathematical procedure to represent the analyte concentration in samples falling within a given range when a method exhibits linearity. The confidence interval around the regression line’s slope is commonly used to depict linearity.
Limits of detection and quantitation
The lowest concentration of an analyte in a sample that can be identified but not measured is known as the limit of detection (LOD). A concentration at a specified signal-to-noise ratio, usually 3:1, is used to represent LOD. On the other hand, the limit of quantitation (LOQ) is defined as the lowest analyte concentration in a sample that can be precisely and satisfactorily measured under the method’s specified operating parameters. The ICH recommends a signal-to-noise ratio of 10:1 for LOQ. The standard deviation of the response (SD) and the slope of the calibration curve(s) at concentrations near the LOD can also be used to calculate both LOD and LOQ, as shown by the calculations below.
LOD = 3.3 × S /SD and LOQ = 10 × S /SD
Specificity
The ability to clearly distinguish the analyte from potentially present components is known as specificity. These elements usually consist of the matrix, degradants, and contaminants, among others. By using one or more supporting analytical procedures, the lack of specificity in a given analytical technique can be minimized. This definition has multiple ramifications: Identification: to verify an analyte’s identification. Purity tests are used to ensure that all analytical techniques, including those involving related chemicals, heavy metals, and residual solvents, enable an accurate declaration of an analyte’s impurity concentration. The purpose of assay (content or potency) is to provide a precise result that permits an accurate pronouncement about the analyte’s content or potency in a sample.52
Range
The range of the method is the distance between the analyte’s upper and lower levels that have been determined with a satisfactory degree of linearity, accuracy, and precision. This range is usually expressed in the same units as the test findings and is derived from either a linear or nonlinear response curve (i.e., when many ranges are involved, as shown below).53
Robustness
In medication development, forced degradation is essential because it determines degradation pathways and evaluates the stability of therapeutic products. This procedure helps with understanding the chemical characteristics of drug molecules, developing stability-indicating techniques, and formulating stable products. For characteristics that are likely to change while being stored, the ICH guideline Q1A emphasizes the significance of using proven stability-indicating testing techniques. Research on forced decomposition should be conducted at pH extremes, at 10°C increments, and under oxidative and photolytic circumstances in order to identify intrinsic stability traits and degradation routes.
Applications
Drug design and the evaluation of the quality, stability, and safety of pharmaceutical compounds—whether synthesized, obtained from natural sources, or produced through recombinant techniques—have seen a wide range of applications. Alkaloids, amines, amino acids, analgesics, tranquilizers, antibacterials, anticonvulsants, antidepressants, antineoplastic agents, macromolecules, steroids, and other compounds are all used in these applications.
Conclusion
The contaminants present in drug substances and drug products are discussed in this article. Drug safety is getting more attention in the literature, and pharmaceutical impurity profiles are becoming more and more important. These days, identifying the contaminants in active pharmaceutical ingredients (APIs) and final drug products is required by a number of pharmacopoeias. Impurity profiling might therefore be a crucial technique for quality control. Important details about the toxicity, safety, detection limits, and quantitation limits of many organic and inorganic contaminants that commonly accompany APIs and final products can be obtained from it. Unique norms and requirements pertaining to contaminants are urgently needed.
Acknowledgement: The authors are thankful to the Guide, Principal, management and Librarian.
Conflict of interest: NoFunding: Not applicable
Ethical consideration: Not applicable
About the Authors
Ms. Priyanka Suresh Ghugare is a researcher in Pharmaceutical Sciences at NIMS College of Pharmacy, NIMS University, Jaipur. Her current academic focus centers on drug formulation, novel drug delivery systems, and excipient characterization. She is actively involved in laboratory research and has contributed to ongoing projects in formulation optimization, analytical method development, and process validation. Through her work, she aspires to advance pharmaceutical methodologies that improve drug efficacy and quality.
Dr. Sandeep Kumar is an Associate Professor in the Department of Pharmaceutics at NIMS College of Pharmacy, NIMS University, Jaipur, with over 9 years of experience in pharmaceutical research and teaching. His expertise lies in the development of targeted drug delivery systems, including nanoparticles, liposomes, and other advanced drug carriers aimed at improving bioavailability and therapeutic efficacy. Dr. Kumar has published more than 40 research articles in peer-reviewed journals and has been an active contributor to various international conferences in the field of pharmaceutics. His research interests also include controlled-release systems, formulation stability, and the biopharmaceutical aspects of drug delivery. He continues to mentor graduate students, guiding them through complex research projects, and has made significant contributions to the academic development of pharmaceutics education at his institution.